Reversible protein phosphorylation occurring on serine, threonine and tyrosine in eukaryotes, one of post-translati-onal modifications (PTMs), plays a crucial role in regulation of the many cellular processes including signal transduction, differentiation, transcription and translation.1-4 Aberrant phosphorylation in cell signaling pathway can lead to mutation, overexpression or abnormal operation of regular enzymes such as protein kinases and phosphatases.5,6 Hence, the global profiling and quantification of phosphoproteins and their phosphorylated sites are of importance for understanding the biological pathway related with the key biological processes in a cell.7-9
Liquid chromatography-mass spectrometry (LC-MS) have been reported for analyzing the phosphoproteins and their phosphorylation sites because of the high sensitivity, precision and accuracy.10,11 Especially, tandem mass spectrometry (MS/MS) has served for information associated with the presence of phosphorylation events and their localization on phosphoproteins. However, the detection of phosphoproteins by MS might be difficult because phosphorylation of proteins is a transient modification, phosphoproteins within cells are relatively low stoichiometry in comparison to nonphosphorylated counterparts, and phosphate group causes by ionization suppression in MS analyses.12,13 Thus, the fractionation and enrichment strategies prior to MS analysis are necessary for reducing the above mentioned challenges.
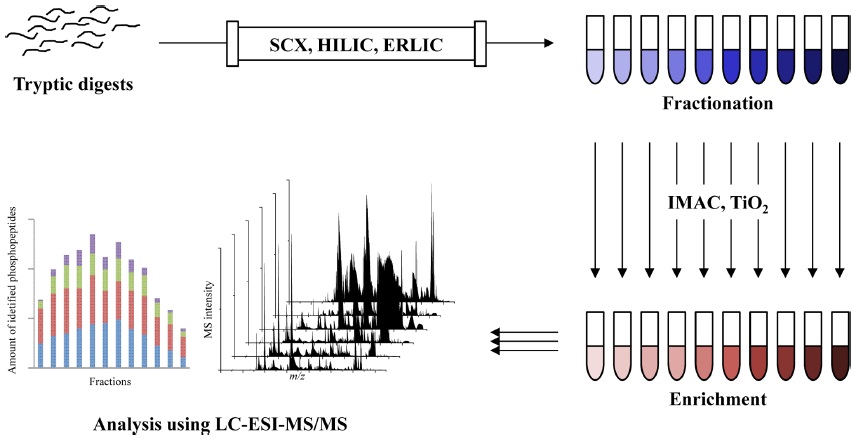
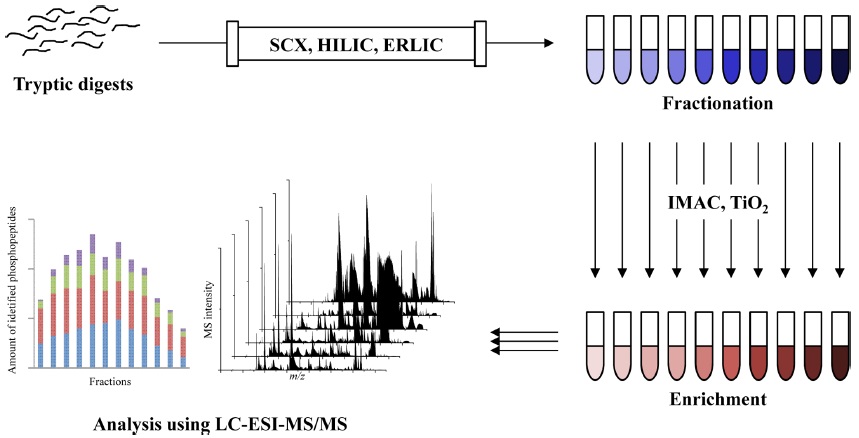
A variety of enrichment and fractionation methods in phosphoproteomics have been reported including the enrichment with immobilized metal affinity chromatography (IMAC)6,14-16 and metal oxide affinity chromatography (MOAC) as well as titanium dioxide (TiO2),17-25 and fractionation with strong cation exchange (SCX),26,27 hydrophilic interaction chromatography (HILIC),28-30 and electrostatic repulsion-hydrophilic interaction chromatography (ERLIC).31-33 More recently, the two sequential steps of preparation methods are commonly used in large scale phosphoproteomics. A fractionation step performs to reduce sample complexity, and followed by an enrichment for increasing the number of identified phosphopeptides and their phosphorylation sites, such as SCX-TiO2,34-36 SCX-IMAC,37,38 ERLIC-IMAC39 and IMAC-TiO2.40,41 Overall workflow of phosphoproteomic analysis using fractionation and enrichment are illustrated at Figure 1.
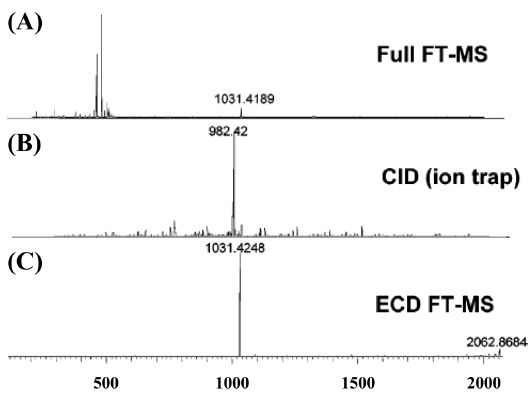
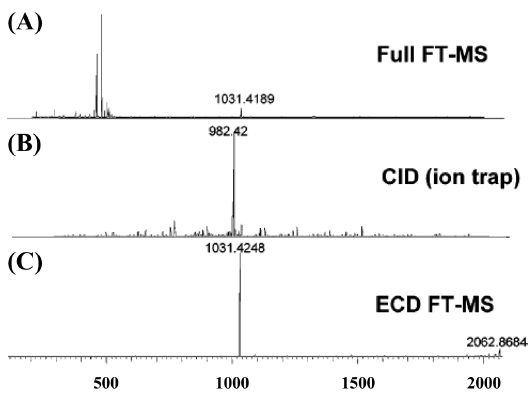
MS/MS-based experiments of phosphoprotein is accomplished with the fragmentation techniques, such as collision induced dissociation (CID),42-44 electron capture dissociation (ECD),45 and electron transfer dissociation (ETD).46 Phosphoproteins consisting of phospho-serine and phospho-threonine residues in MS/MS analysis with CID are often produced the significant neutral loss of phosphate group, and these phenomenon affects to fragmentation for identification of phosphoproteins. On the other hand, phosphoprotein analysis by ECD or ETD prevents to cleave the unstable phosphate group, and offers the unambiguous information of phosphorylation site locations.47
This article provides an overview of preparative and analytical strategies using LC-MS/MS for identification and characterization of phosphoproteins and their phosphorylation sites localizations. Furthermore, we reviewed on the fragmentation reactions of phosphoproteome by MS/MS in understanding biological network pathway.
>
Strong cation exchange (SCX )
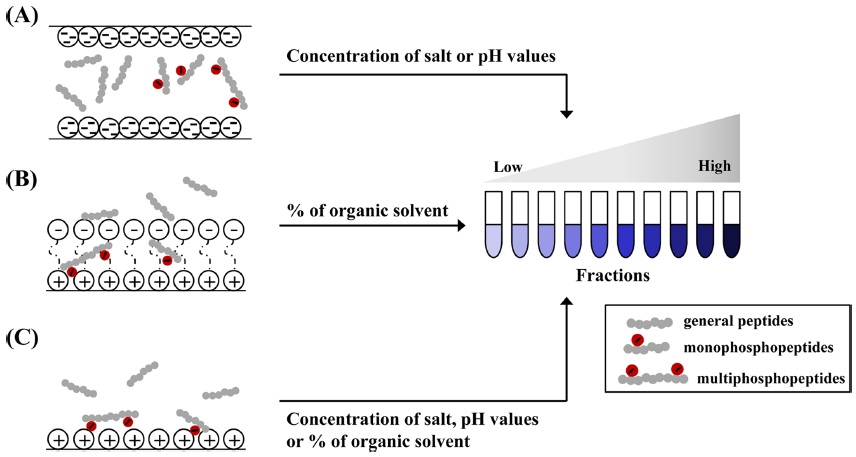
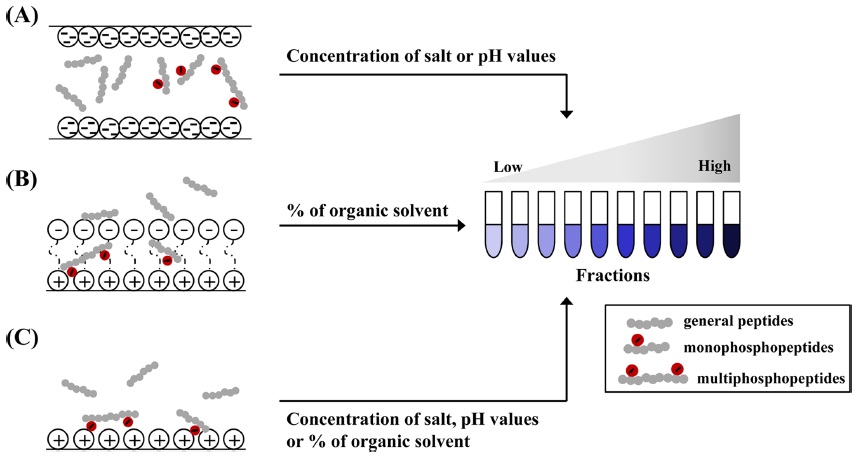
SCX chromatography is a universal fractionation technique for phosphoprotein (or phosphopeptide).48,49 Proteins or peptides are retained on a column that contains a hydrophilic, anionic resin through the affinity to positively charged groups on amino acids (Figure 2-A). The main principle of SCX is the relationship between SCX matrix and oppositely charged proteins (or peptides) from biological sample according to their net surface charge and
>
Hydrophilic interaction chromatography (HILIC)
HILIC for separating the proteins and peptides rely on the partition of analytes between the used solvents and solid resins.29 In HILIC, the hydrophilic peptides take to remain into chromatography column, the hydrophobic peptides eluted at early step in contrast with reverse phase chromatography. HILIC has been reported frequently in recent years in phosphoproteomic studies, because highly hydrophilic solutes as phosphorylated ones in comparison with common peptides are retained on the column for the longest time.28-30 In HILIC, hydrophilic components acquired from biological specimens are typically bound onto HILIC column matrix when used with below 10 % aqueous. To isolate the phosphopeptides from ordinary ones, all peptides are fractionated by predominantly increasing the aqueous mobile phase up to 40% (Figure 2-B). Fractionations using HILIC facilitates the identification and characterization of phosphopeptides and number of phosphorylation sites, when combined with either IMAC or TiO2 . Zappacosta and co-worker provided a collaboration of HILIC and IMAC for identification of more than 20,000 phosphopeptide from rat liver.30 Most non-phosphopeptides from rat liver were removed in early steps (fraction 1~15), followed by the elution of mono and multiphosphopeptides by stepwise. Accordingly, HILIC is an alternative tool for phosphoprotein or phosphopeptide fractionation, and thus can be coupled online to MS analysis.
>
Electrostatic repulsion-hydrophilic interaction liquid chromatography (ERLIC)
ERLIC chromatography, one of phosphopeptide fractionations, was first introduced by Alpert in 2008.31 This technique is modified from HILIC approach and can be accomplished isocratically.32 ERLIC matrix containing weak anion exchange and hydrophilic interactions allows for the isolation of phosphopeptides from other ones in proteolytic digests. Many phosphorylated proteins and peptides are more hydrophilic in comparison with their unmodified counterparts, can be separated with ERLIC (Figure 2-C). Separation of ERLIC is influences by pH, salt concentration, count-ion, a percent of organic solvent in mobile phase, etc.32,33 ERLIC matrix prior to sample loading are acidified with a combination of organic acids with high percent of acetonitrile to remain the phosphopeptides from tryptic digests, and gradually fractionated by increasing concentration of salts. The eluted phosphopeptides using ERLIC protocol is recently applied in combination with reverse phase, TiO2,50 SCX,32,33 and IMAC.39 Loroch, S. and co-worker demonstrated that HeLa cells digests were sequentially separated to 21 fractions using ERLIC combined with SCX, total 8998 phosphorylation sites from 3013 phosphoproteins are detected.32 Early elutions (fraction 1-5) among 21 fractions are detected with mono-phosphopeptides, whereas the pooled middle fractions (fraction 10-15) are confirmed with multi-phosphopeptides.
>
Immobilized metal ion affinity chromatography (IMAC)
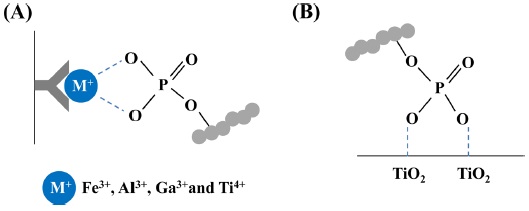
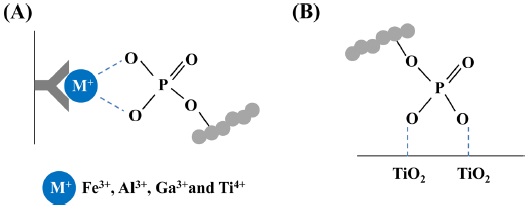
IMAC is a widely used technique to enrich phosphopeptides prior to MS analysis. Positively charged metal ions, such as Fe3+,16,47 Zn2+,15 Ga3+,51 La3+,52 and Ti4+,38,47 are chelated to nitrilotriacetic acid (NTA) or iminodiacetic acid (IDA) resin and interacted with negatively charged phosphate groups (Figure 3A).One of the limitations in the IMAC technique is its partial non-specific binding of acidic peptides containing glutamic acid or aspartic acid residues when used for phosphopeptide enrichment, thereby introducing the inefficiency of IMAC. The pH values in IMAC specificity are an important factor, because the acid dissociation constant (pKa) of phosphate group at serine/threonine (pKa 2.1) and tyrosin residues (pKa 1.0) is lower than other acidic amino acid including glutamic acid (pKa 3.65) and aspartic acid (pKa 4.25).53-55 To minimize these issues, loading and binding buffers during phosphopeptide enrichment with IMAC were often used to pH 2-2.5 containing with organic acid [
The TiO2 -based strategy, one of metal oxide affinity chromatography (MOAC), is the robustness, simplicity, and high efficiency for enrichment of phosphorylated proteins or peptides from complex biological sample (Figure 3B). TiO2 techniques are usually performed using self-packed or commercial products with microcolumn,17,56 or beads.20,25,40 To improve the reproducibility of TiO2 for phosphopeptide enrichment, Tape and co-worker described that the automatic phosphopeptide enrichment using magnetic TiO2 beads with IMAC.40 The automated enrichment system with magnetic TiO2 beads is achieved by controlling 96-well plates at the eight carousel positions. Phosphopeptides from tryptic digests are more identified with magnetic beads and automatic system, compare to manual enrichment.
Selectivity of TiO2 resin is influenced by the pH value of loading and eluting buffers similar to IMAC approach. To enhance the enrichment efficiency of TiO2 resin, the loading buffer was acidified by organic acids (
In addition, on-line TiO2 -based approach reported for the reproducibility, use-to-easy, and high-throughput in phosphoproteomics, unlike the abovementioned off-line approaches.55,62,63 Pinkse and co-worker demonstrated that online phosphopeptide enrichment with TiO2 resin is automatically carried out using ‘sandwich precolumns’ made up of reverse phase (RP)-TiO2 -RP materials.55 All peptides obtained from tryptic digests are remained in first RP precolumn at 3 µL/min. During an initial H2O/ACN gradient at low flow rate (~100 nL/min), non-phosphopeptides and phosphopeptides are transferred to analytical column and TiO2 precolumn. The phosphopeptides binding on TiO2 resins are released by injecting the elution buffer at high pH value and concentration of salts in a second H2O/ ACN gradient. Briefly, pH value of buffer solution, concentration of salts, and flow rate are the crucial parameters for phosphopeptide enrichment using online RP-TiO2-RP system. The above processes on TiO2 protocol are beneficial for increasing the probability of phosphopeptide identification.
>
Phosphopeptide entichment using β-elimination reaction
Affinity purification in cooperation with β-elimination of phosphopeptides is helpful for isolating phosphopeptide by modifying phosphate groups.64-66 McLachlin and co-worker introduced the improved chemical modificationbased phosphopeptide enrichment strategy.65 β-Elimination of phosphopeptide can be achieved with the solution made up of 25 mM
>
Combination with fractionation and enrichment
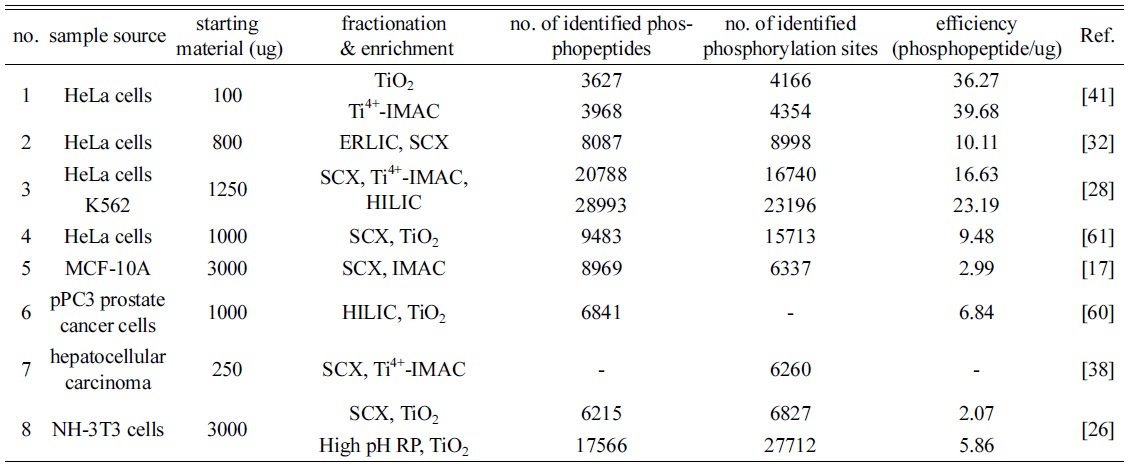
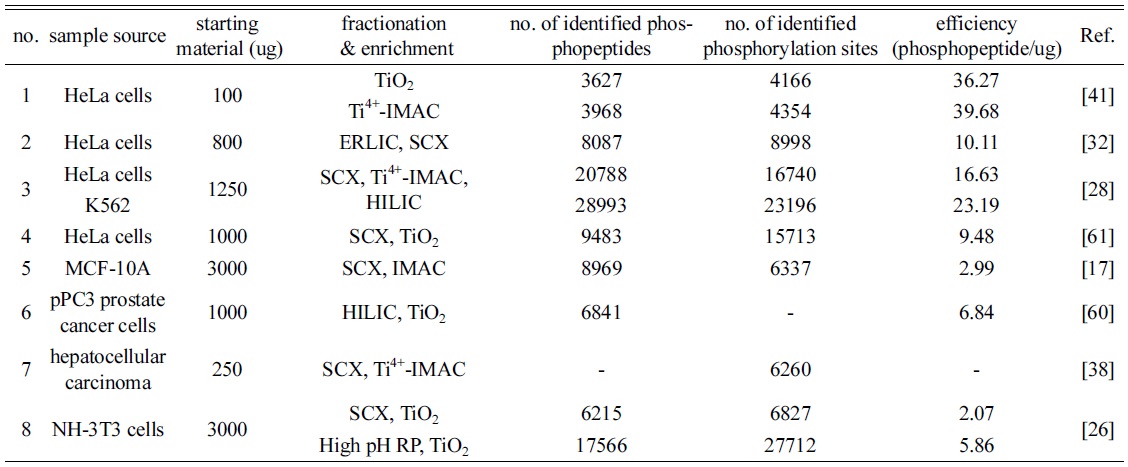
In phosphoproteomics, the two sequential steps with fractionation and enrichment methods are, in some cases, used to more identify phosphopeptides and their phosphorylation sites up to date. As shown in Table 1, the starting materials of biological samples for phosphopeptide enrichment are used at approximately 0.1~3 mg, and the maximum amount is 15 mg of cell lysates. Most combinational strategies achieve utilizing two preparation methods, and present the synergy effect for identifying phosphopeptides and their phosphorylated sites than each approach. Zhou and co-worker introduced 3D-cooperative strategies with SCX, Ti4+-IMAC and HILIC.28 Tryptic digests are fractionated
[Table 1.] The recent trends of fractionation and enrichment in phosphoproteomics
The recent trends of fractionation and enrichment in phosphoproteomics
>
Mass spectrometry analysis of phosphopeptides
Phosphopeptides enriched from tryptic digests are introduced into MS
We have introduced the overview of diverse fractionation and enrichment strategies for isolation of phosphoproteins and phosphopeptides from complex cellular components.
The cited phosphopeptide fractionating and enriching protocols are respectively relied on ion strength and chelating reaction between their matrix and phosphopeptides, and contributing to discovery of low abundant phosphopeptide functions and early diagnosis of diverse diseases. Additionally, the combination of phosphopeptide enrichment with sample fractionation prior to MS analysis is of key importance to overcome the existing challenges and to enable the identification of more phosphopeptides and phosphorylation site localizations.
The entire above-mentioned fractionation and enrichment approaches are well appropriated in phosphoproteomics, and two or more parallel strategies can be got the new information for understanding the function of phosphorylations in cellular network, and thus can help to identify novel phosphoproteins (or phosphopeptides) involved in therapeutic mechanisms from diseases.