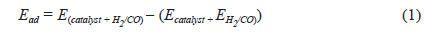Direct alcohol fuel cells (DAFC) have the advantages of high energy density, easy handling of liquid fuel and low operating temperatures, making them a potential candidate system for power generation in portable equipment or devices [1]. Among the various types of DAFCs, direct methanol fuel cells (DMFCs) have been extensively developed as promising devices for the portable power market because of the easy oxidation of methanol [2,3]. When Pt is used as a catalyst on a DMFC anode, the electro-oxidation of methanol (<80℃) shows sufficient catalytic activity to produce H2. Unfortunately, Pt tends to be readily poisoned by carbon monoxide (CO), a reaction by-product, and thus in time becomes deactivated [4]. For this reason, many studies have focused on enhancing the catalytic activity by inhibiting the CO poisoning effect using carbon-supported binary catalysts including Pt, by means of an electronic effect or an ensemble effect [5-13]. Most of this research has focused on the role of the catalyst in maintaining activity. However, it is also possible that the carbon support is not clean enough to maintain sufficient activity.
We investigated the influence of defect sites on the carbon support using density functional theory (DFT) to obtain a fundamental understanding of adsorption characteristics relevant to H2 dissociation and CO poisoning on Pt/C at the molecular level. We also discuss the effect of the defects of a carbon substrate on catalytic activity due to CO poisoning.
In this investigation, DFT calculations were performed to evaluate H2 and CO adsorption on the Pt sites and defect sites of carbon-supported Pt systems, using the three-dimensional periodic slab model. Geometry optimizations were performed using DMol3 (Dassault Systèmes BIOVIA, San Diego, CA, USA) under the following conditions: 1) the spin-polarized general gradient approximation (GGA)-PBE functional [14,15] option, and 2) the DNP basis set, to predict the properties of catalyst and adsorbates (H2 or CO). GGA-PBE based functions have been extensively used to describe systems including Pt catalysts [16-18] and carbon-based materials [19-25].
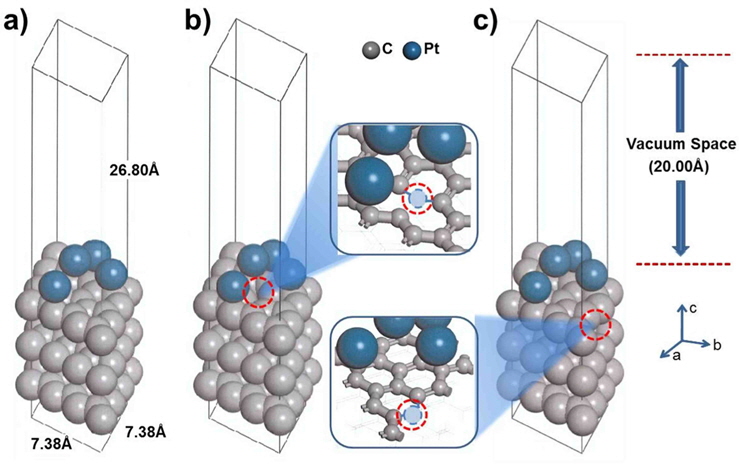
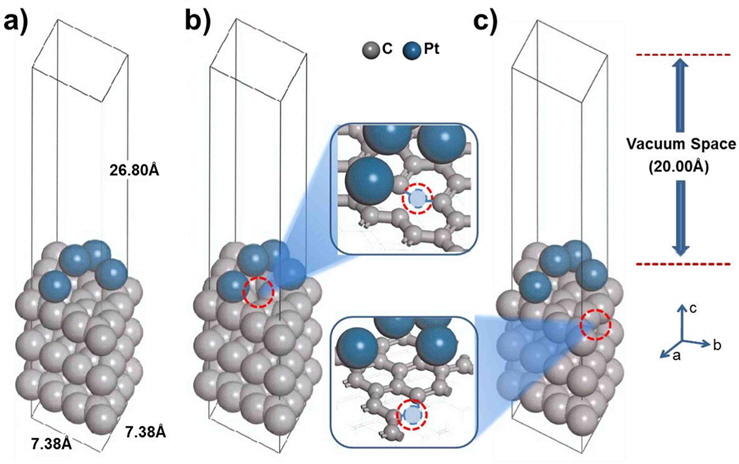
In the supercell, the surface was exposed as a slab, providing four Pt atoms over three layers of carbon (graphite). For the development of the surface structure, three atomic layers of graphite were cleaved at the (001) plane. The vacuum thickness (size of the unit cell perpendicular to slab-slab thickness) was set to 20 Å to prevent interactions beyond the periodic boundary. The Brillouin zone was sampled using the 4 × 4 × 1 Monkhorst-Pack (MP) k-point mesh [26]. The self-consistent field (SCF) convergence, 1 × 10−5 Ha, was obtained at the given k-point sampling. Four Pt atoms [27,28] were loaded on graphite, and the system was optimized with a constraint of the third layer, as shown in Fig. 1. We also designed a mono-vacancy carbon substrate to study the effect of defect sites, where Pt is bound to a neighboring site (nbr-Pt/C) or a distant site (dis-Pt/C) on the carbon (001) surface, as shown in Fig. 1b and c.
To determine adsorption energies
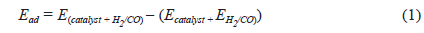
where Ead is the adsorption energy of the adsorbate on the adsorbent;
3.1. H2 and CO adsorption on Pt/C
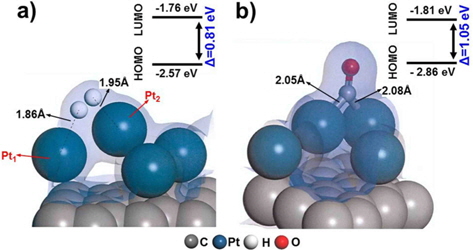
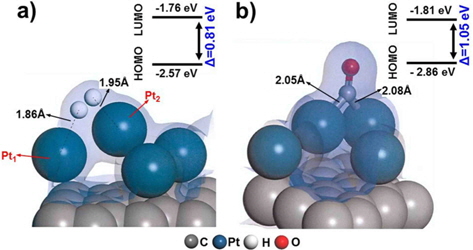
We first evaluated the adsorption of H2 and CO on a “clean” Pt/C surface i.e., a surface with no defect sites. The geometry of H2-Pt/C and CO-Pt/C was optimized to identify the preferred molecular conformation, as shown in Fig. 2. Table 1 shows that H2 and CO were strongly adsorbed on Pt, with adsorption energies of −1.38 eV and −2.19 eV, respectively. This is in good agreement with the findings of previous theoretical (−1.4 eV and −2.17 eV) and experimental studies [5,6,29,30]. The electron density field also supports the enhanced electronic interaction between Pt and H2/CO. H2 was readily dissociated on Pt clusters to form Pt1-H and Pt2-H bonds, which is typical for H2 dissociation on Pt [30]. H2 splits into two hydrogen atoms, and each bonds to a Pt atom: one with a distance of 1.86 Å, and the other with a distance of 1.95 Å. CO is also favorably bonded in a bidentate fashion because of the highly interactive electron field of CO-Pt, which confirms that CO poisoning is favorable within Pt clusters.
[Table 1.] Adsorption energies of H2/CO on catalyst systems

Adsorption energies of H2/CO on catalyst systems
3.2. H2 and CO adsorption on nbr-Pt/C
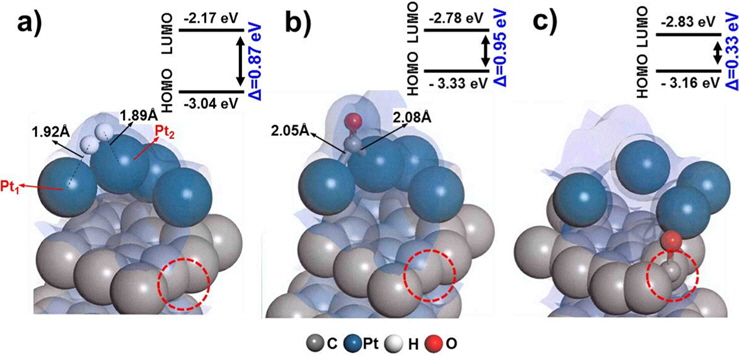
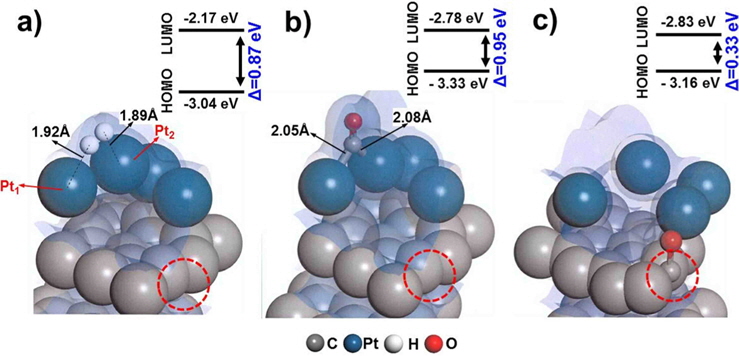
Defect surfaces on Pt/C anodes play a notable role in the anode activity, as increased interaction with CO can lead to poisoning and oxidation of the carbon surface. To probe the effect of defect sites, we first investigated H2 dissociation and CO adsorption on Pt/C, where the defect site is located neighboring to a Pt cluster (nbr-Pt/C), as shown in Fig. 3a. Interestingly, for the adsorption of both H2 and CO, one Pt atom (Pt1) filled the neighboring defect site, sharing electron density with the defective carbon surface. Meanwhile, the reduced metal content resulted in diminished H2 dissociation, as shown by the smaller adsorption energy (−1.09 eV) and H-H distance (0.80 Å) compared to those of clean-Pt/C (−1.38 eV and 0.88 Å, respectively).
CO adsorption was expected to be reduced in a similar manner, but the adsorption increased significantly, as shown by the large bond strength (−3.02 eV) and bond length of CO (2.06 Å). The electron density field shows that density is shared between Pt-CO and carbon. Considering the molecular orbitals during the reaction, the degree of electron delocalization of the system leads to a change in the highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) band gap, which is dependent on the adsorption of the different gas molecules.
Therefore, we investigated the energy trends on the basis of molecular orbitals in order to elucidate the gas binding mechanism. We carried out additional calculations of the electronic energy of the bands and the localization of molecular orbitals for the interaction of H2/CO with the Pt atom. The relatively large band gap for H2 dissociation (0.96 eV) on nbr-Pt/C as compared to that for clean-Pt/C (0.81 eV) suggests that the Pt-H2 interaction on the system with defects was reduced, which is in line with the reduced adsorption energy. Conversely, the band gap for CO adsorption in nbr-Pt/C narrowed (0.95 eV), because of the strong electronic interaction between CO and Pt, even with one Pt atom filling the defect site. The strength of the interaction between Pt-C and CO contributes to the degree of CO chemisorption. The geometry of the dimer Pt-CO bond is consistent with the orbital sharing conformation, because of the Pt-CO* π-back bond donation [30]. Thus, H2 dissociation tends to take place on clean-Pt/C rather than on nbr-Pt/C, where CO poisoning occurs.
3.3. H2 and CO adsorption on dis-Pt/C
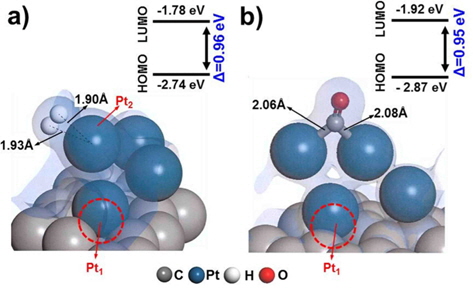
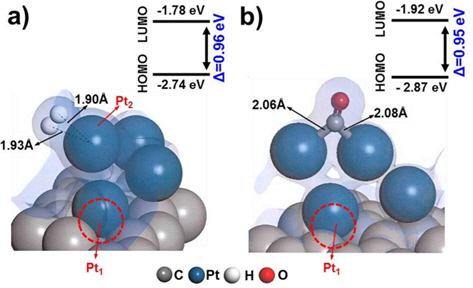
Since defect sites on Pt/C are located away from Pt over the carbon surface, we additionally designed a structure containing a mono-vacancy site distant from the Pt clusters, and then optimized the structure with adsorbed H2/CO molecules, as shown in Fig. 4. Both H2 and CO were strongly adsorbed on Pt with adsorption energies of −1.31 eV and −1.79 eV respectively, which are comparable to the adsorption characteristics on the cleanPt/C surface (Table 1). The electron density field also shows the well-distributed electronic interaction between Pt and H2/CO.
Interestingly, CO perfectly filled the defect site on carbon, with a high adsorption energy (−4.24 eV). CO is preferentially bonded to the carbon of graphite via π bond formation, as CCO-Cgraphite, because of the highly interactive electron sharing of CCO-Cgraphite, which in turn is attributed to the narrow HOMO-LUMO band gap (0.33 eV). CO adsorption on the Pt cluster (0.95 eV) tends to be similar to that on clean-Pt/C (1.09 eV), which represents the independence of the interaction when the defect site is far from the adsorption site. In addition, the oxygen atom of CO causes electro-oxidation of the carbon surface, suggesting that defect sites lead to further CO poisoning. When filled with CO, the matrix could be saturated with H+ to form oxidation side products such as methoxy group products (CxHyOz). CO poisoning deactivates the catalytic performance on an anode surface having defect sites. From the results of DFT calculations, it is noteworthy that defect sites on carbon significantly increased CO poisoning in the DMFC anode.
We investigated carbon-supported Pt catalyst systems as DMFC anodes with defect adsorption sites, in order to elucidate the mechanisms of H2 dissociation and CO poisoning, using quantum mechanical DFT methods. We found that neighboring Pt/C defect sites suppressed H2 dissociation and CO poisoning due to Pt atoms filling the defect sites. With distant Pt/C defect sites, H2 dissociation was still active on Pt, but CO could fill the defect sites to form carbon π-π bonds, thus enhancing the oxidation of the carbon surface. In addition, the oxygen atom of CO caused electro-oxidation of the carbon surface, suggesting that the presence of defect sites can lead to CO poisoning. Therefore, defect sites on the carbon support significantly affect CO poisoning on the DMFC anode.