Protease, renowned as proteolytic enzymes or proteinases, refers to a group of enzymes that hydrolyzes (breakdown) proteins into small peptides and amino acids. Proteolytic enzymes are essential in various therapeutic purposes such as oncology, inflammatory conditions, blood rheology control, and immune regulation. Undigested proteins, cellular debris and blood toxins can also be digested by proteases. Current classification of enzymes unveils six broad groups of proteases, i.e. serine proteases, threonine proteases, cysteine proteases, aspartate proteases, glutamic proteases and metalloproteases. Alternatively, on the basis of the isoelectric points (pI) of catalytic proteins they can also be classified into acid, alkaline and neutral proteases. Acid and neutral proteases are involved in type I hypersensitivity by activating complement systems and kinins (Mitchell et al., 2007) and the function of alkaline or basic proteases of
Previous
Present work has been designed to understand the natures of different types of
A total of 84 acid, 283 neutral and 31 alkaline proteases of
>
Physiochemical parameters analysis
Physicochemical data were generated from ProtParam software using ExPASy server (the proteomic server of Swiss Institute of Bioinformatics). FASTA sequence format were applied for subsequent analysis.
>
Protein superfamily and family search
The Superfamily tool on ExPASy server was used for protein family search.
>
Multiple sequence alignment (MSA)
The program ClustalW2 (Larkin et al., 2007) was used for multiple sequence alignment and MSA was represented by CLC-Bio sequence viewer.
>
Phylogenetic tree construction
Phylip-3.69 (Tuimala, 1989) was used for phylogram construction by Neighbor-joining (NJ) method using 100 bootstrap values. Tree was edited by Dendroscope (Huson et al., 2007).
Selected protein sequences were studied for protein-protein interaction to detect the probable function using STRING Database.
Acid and neutral proteases were separately subjected to Pfam to find out conserved domains. Separated domains were subjected to Block Maker for conserved block identification. Separated blocks were used for motif finding using MEME Suite. Conserved motif of alkaline protease was deduced from the multiple sequence alignment.
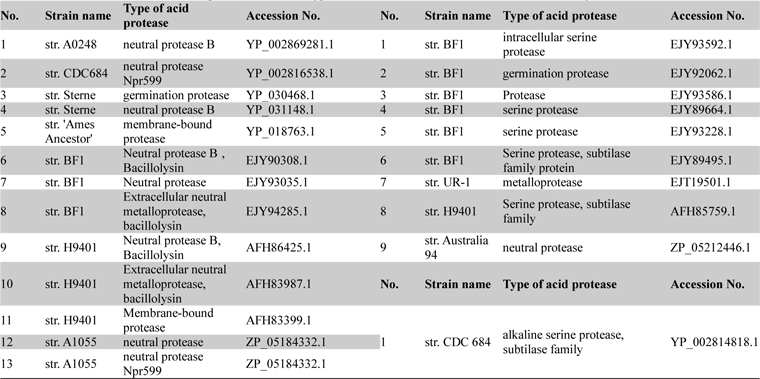
Among all the deposited
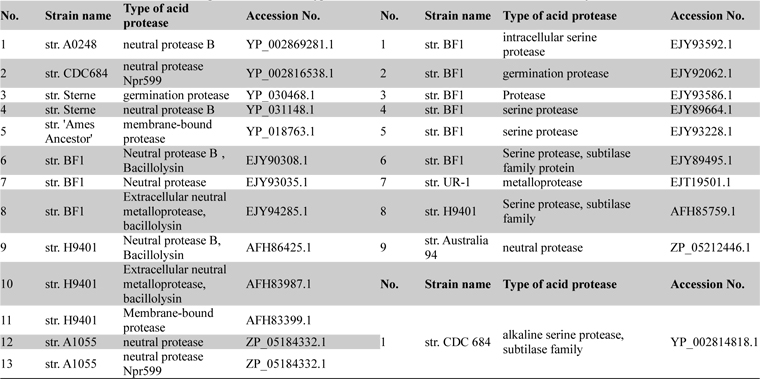
List of acid neutral and alkaline proteases with their type and accession numbers which were taken for analysis
>
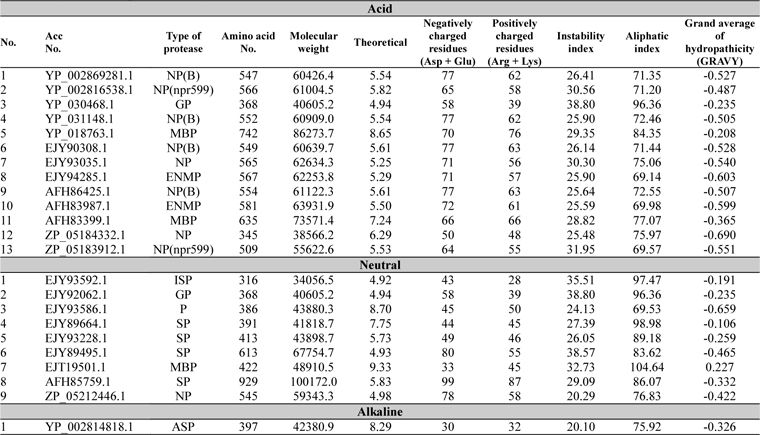
Physicochemical parameter analysis of proteases
The physicochemical features of protease sequences were represented in Table 2. The amino acid number for acid protease ranged from 345 to 742 with variable molecular weight. The pI value varied from 4.94 to 6.29, except sequences 5 and 11 (Acc. No. YP_018763.1 and AFH83399.1), which have their pI of 8.65 and 7.24 respectively. The above mentioned two sequences (5 and 11) were membrane bound proteases which have the aliphatic index value of 84.35 and 77.07 respectively. On the other hand sequence 3 (Acc. No. YP_030468.1) has the highest aliphatic index value of 96.36.
[Table 2.] Physiochemical parameter analysis
Physiochemical parameter analysis
For all the neutral proteases group of protein different range were found in different analysis (Table 2). Germination protease (sequence 2) showed all the values similar to that of the germination protease of acid protease group (sequence3) with 4.94 pI value. Some serine proteases were also found in this group with various pI values. Accession number EJT19501.1 (Sequence 7) which was a membrane bound protease, showed the highest pI value of 9.33 and aliphatic index value of 104.64.
>
Protein superfamily and family search
The entire sequences of acid, neutral and alkaline proteases when subjected to Superfamily tools on ExPASy server revealed different superfamily and family (Table 3). For acid protease 10 sequences were found with Metalloproteases (''zincins''), catalytic domain superfamily and Thermolysin-like family (Table 3). Sequence 3 (Acc. No. YP_030468.1) was found with HybD-like superfamily and Germination protease family. Sequence 5 and 11 (Acc No. AFH83399.1 and YP_018763.1) were found with Cysteine proteinases superfamily and Transglutaminase core family. The short segments were found to have similarity with thermostable phytase (Table 3). But in the case of neutral proteases most variable domains were observed in superfamily and family analysis (Table 3). Among them sequence 7 and 9 were related to ten acid proteases, sequence 2 showed similarity with germination protease sequence 3 (acid protease) and sequence 8 specified similarity with alkaline protease 1. So a sequence level dissimilarity was observed among 9 neutral proteases. These have been reflected in the multiple sequence alignment also.
Distribution of superfamily and family among acid, neutral and alkaline proteases of Bacillus anthracis
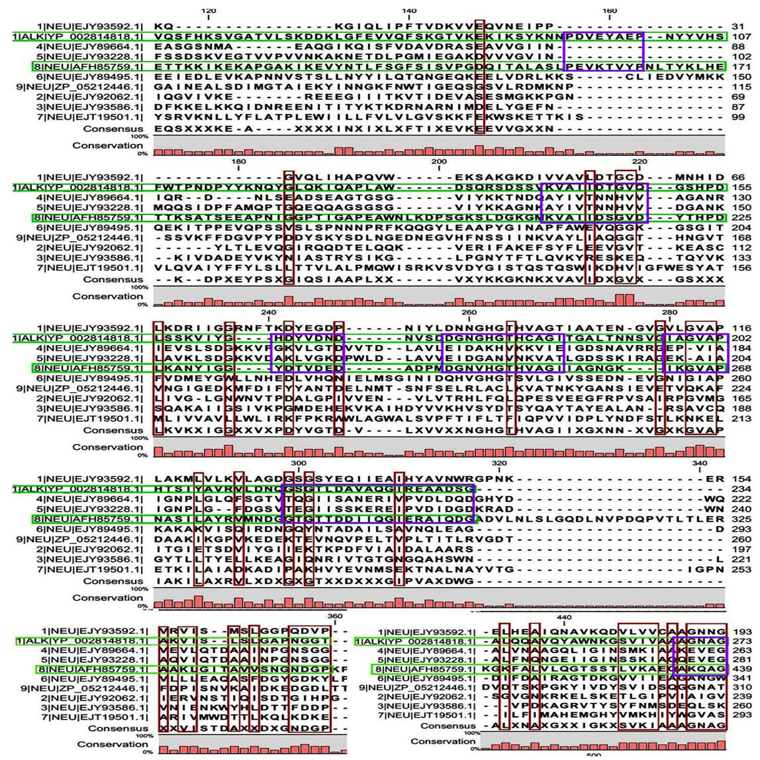
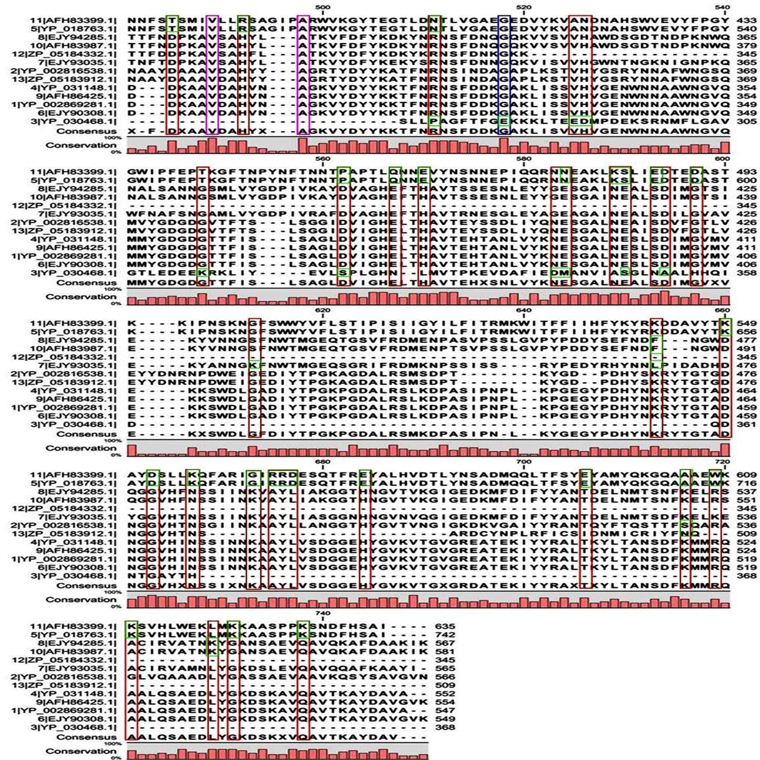
Multiple sequence alignment analysis of the 13 acid proteases, the 9 neutral proteases and the one alkaline protease displayed the superfamily results of each groups. Fig. 1A and B showed consensus regions of acid proteases. Presence of consensus regions throughout the whole alignment indicated high level of sequence similarity among them. Three 100% conserved positions were found in aligned region such as position 368, 489 and 498 which have been represented in pink bar. Blue bars represented some specific changes only for sequence 3 (germination protease). Red bars represented near about conserved regions where green bars indicated the changes. In maximum cases, changes were found for two membranes bound proteases sequences 5 and 11 (Accession No. YP_018763.1 and AFH83399.1) and germination protease sequence 3 (Accession No. YP_030468.1).
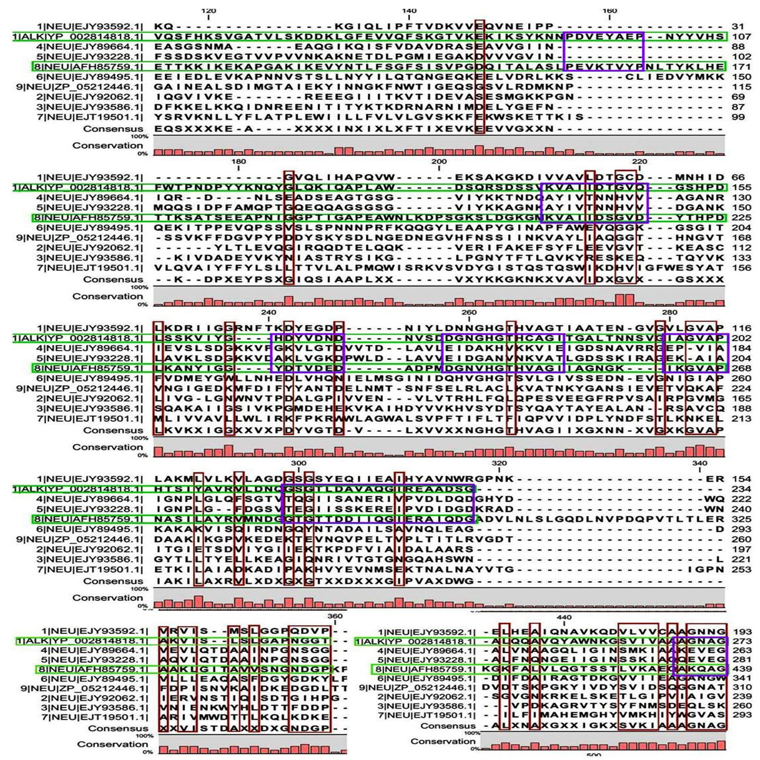
A few ranges of consensus regions were found with low levels of sequence similarity in multiple sequence alignment of neutral protease group and one alkaline protease. Red bars indicated the similarity area. As the alkaline protease sequence (Accession No. YP_002814818.1) showed highest similarity with neutral protease sequence 8 (AFH85759.1) in previous experiments, they have been presented in green colour (Fig. 1C and D). Ten short conserved motifs of alkaline protease were also showed in violet box in comparison with neutral protease 8.
>
Phylogenetic tree construction
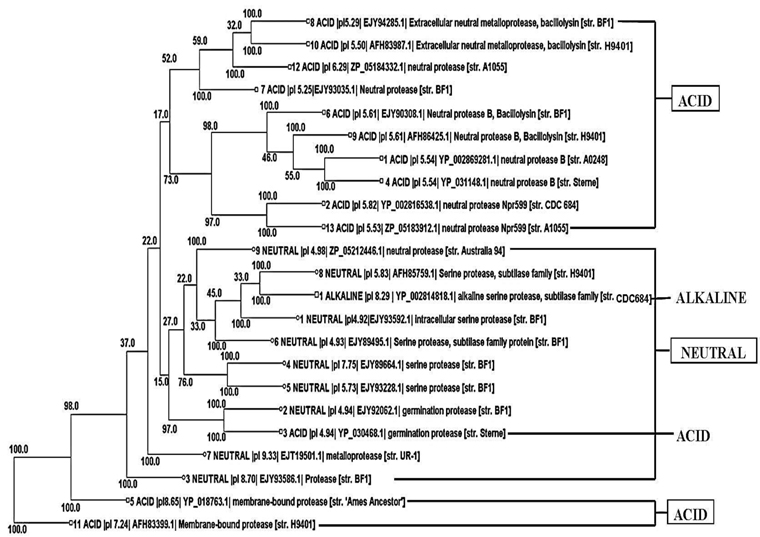
Phylogenetic tree construction of all the 13 acid protease, the 9 neutral protease and the one alkaline protease showed an interesting result. It was found that 10 acid protease were cluster together in the top of the tree (Fig. 2). Two membranes bound protease sequences 5 and 11 (YP_018763.1 and AFH83399.1) were found together in the bottom of the tree. Germination protease sequence 3 was found with another germination protease sequence 2 of neutral protease group. One alkaline serine protease was found with the neutral serine protease sequence 8.
Protein-protein interactions are the core of the interactom study which also represents the secretom of an organism. Here in this study the interaction of acid, neutral and alkaline protease of
A total of 6 motifs were found from acid and neutral protease (Table 4). Motif A2 and A3 showed similarity with peptidase M4 function as per the BLAST and PFAM result. According to PFAM and GENE3D motif B1, 2, and 3 all have the peptidase activity. The function of B3 deduced by BLAST was endopeptidase spore protease Gpr. Ten short conserved motifs of alkaline protease were identified from multiple sequence alignment (Fig. 1C and D).
Identified motifs for acid and neutral proteases with their function deduced by protein BLAST and INTERPROSCAN. Accession number, query coverage, e-value and maximum identity of highly similar sequence are represented here
The present study reported that
Physicochemical nature of a protein can be easily calculated through
All the metalloproteases of acid group were found to prefer extracellular medium according to GRAVY results (Table 2) and they were moderately thermostable in nature. In reference to the above parameters protein sequences of 5 and 11 were highly thermostable and membrane bound proteases of
Phylogenetic tree (Fig. 2) visibly reflected the superfamily results (Table 3). Although all the proteins were sequentially different, 10 acid proteases showed close relationship according to their evolution. They all showed same domains in superfamily search indicating their similar function. Functional similarity was also found between two germination proteases and also between two membrane bound proteases. The studied neutral proteases were found together in the tree. Among them neutral serine protease 8 showed highest similarity with alkaline serine protease one (Fig. 2), indicating their functional similarity. From the above result it was found that same sequence represented same functional domain and on the basis of that they showed evolutionary relationships.
According to Baillie (2001) and Russell et al. (2007) anthrax pathogen initiates their germination as well as infection by interacting with the host macrophages. The resulting vegetative cells spread in blood and other tissues as the causative agent of meningitis and ultimately causes death. Literature showed that among the secreted metalloproteases of
List of protein-protein interaction study. Accession number, name and functions of interacted proteins were listed here
As 100% sequence similarity were found among 31 retrieved alkaline proteases, it can be concluded that
The