Animal mitochondrial genomes (mitogenomes) are approximately 16~20 kbp, and are encoded with a remarkably conserved set of 37 genes: 13 protein-coding genes (PCGs), two ribosomal RNA (rRNA) genes, and 22 transfer RNA (tRNA) genes, and one major non-coding sequence, which is termed the control region (Boore, 1999). This control region in insect instead is called as the A+T-rich region due to the high adenine and thymine (A/T) content, and in fact, this region contains the highest A/T content of any region of the mitogenomes in insects (Kim et al., 2010).
The mitogenome information has greatly been devoted to our understanding of several fields of biology (i.e., comparative and evolutionary genomics, molecular evolution, and phylogenetics). However, still newly sequenced insect mitogenome information provides us with new insights into genomic structures (Wan et al., 2012) and the evolutionary relationships of several levels of taxonomic groups (Kim et al., 2011; Cameron et al., 2009), and gene arrangement (Wang et al., 2013).
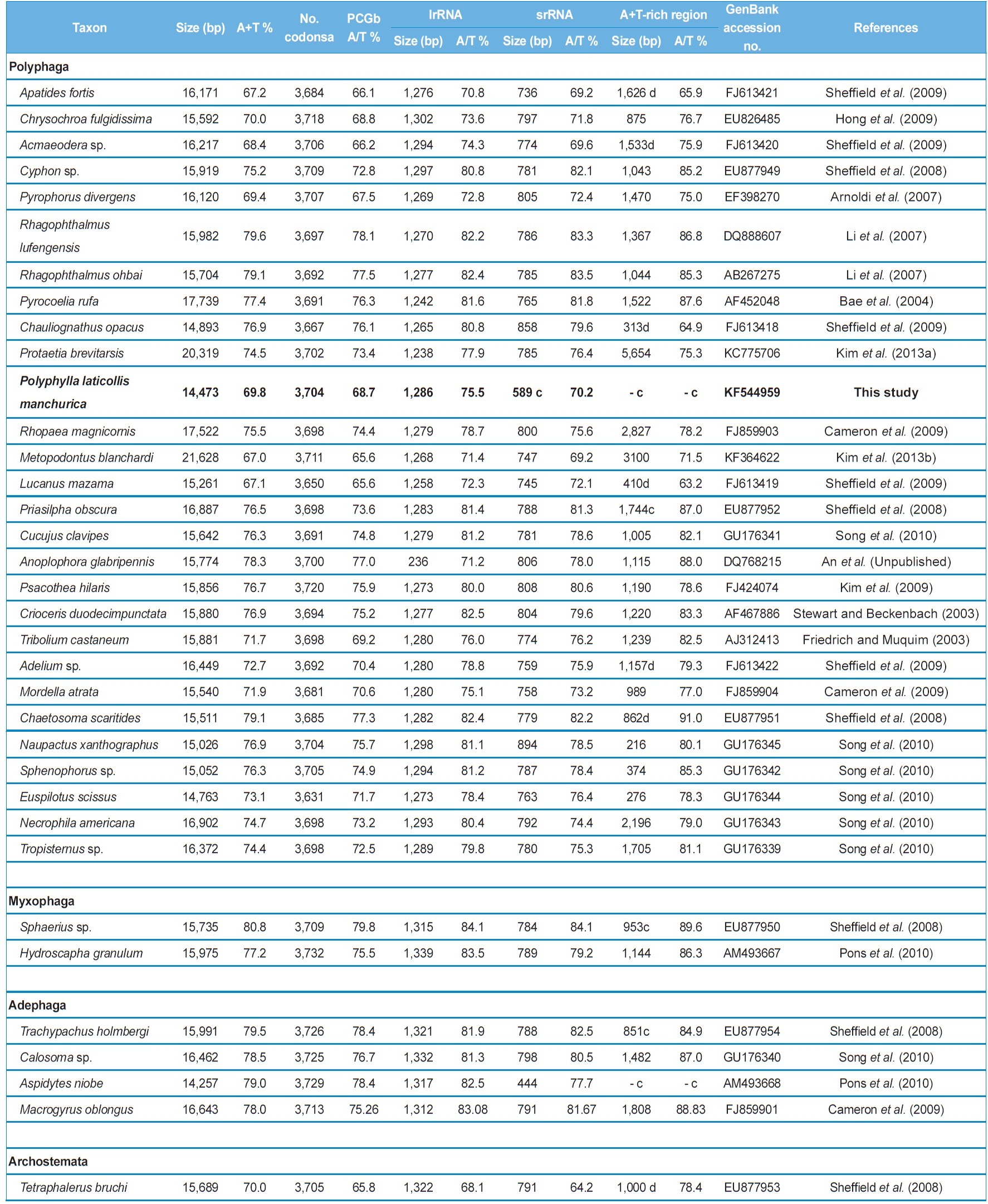
Up to now, more than 250 mitogenome sequences have been determined from a variety of insects, but this list includes only ~34 coleopteran species (http://www.ncbi.nlm.nih.gov/genomes/ORGANELLES). Considering that the suborder Polyphaga contains the vast majority of beetle diversity, with at least 300,000 described species (90% of the beetles) belonging to more than 100 families in four infraorders (Hammond, 1992), genomic information is extremely limited. In particular, the complete mitogenome sequence of the infraorder Scarabaeiformia in Polyphaga is available only for two species (Cameron et al., 2009; Sheffield et al., 2009). Recently, Kim et al. (2013a, 2013b) additionally reported two complete mitogenome sequences of Scarabaeiformia, the whiter-spotted flower chafer, Protaetia brevitarsis (Scarabaeidae) and the two-spotted stag beetle, M. blanchardi (Lucanidae).
In this paper, we report the mitogenome sequence of Polyphylla laticollis manchurica to describe the genome via comparison to those of pre-existing coleopteran insects in terms of whole genome organization, arrangement, and the major characteristics of individual genes. P. l. manchurica is distributed throughout Korea, including Jeju Island, and also in Mongolia and japan, as well as Eastern China (Won et al., 1998). Due to the rarity the species is listed as an endangered species in Korea (Won et al., 1998).
The garden chafer, Polyphylla laticollis manchurica (Scarabaeiformia: Scarabaeidae), was collected in Yeongwol, Gwangwondo-Province in Korea. P. l. manchurica is listed as a first degree endangered species in Korea, and thus, proper permission was obtained from relevant supervisory office before collection. Total genomic DNA was extracted using the Wizard™ Genomic DNA Purification kit (Promega, USA) following the manufacturer's instruction.
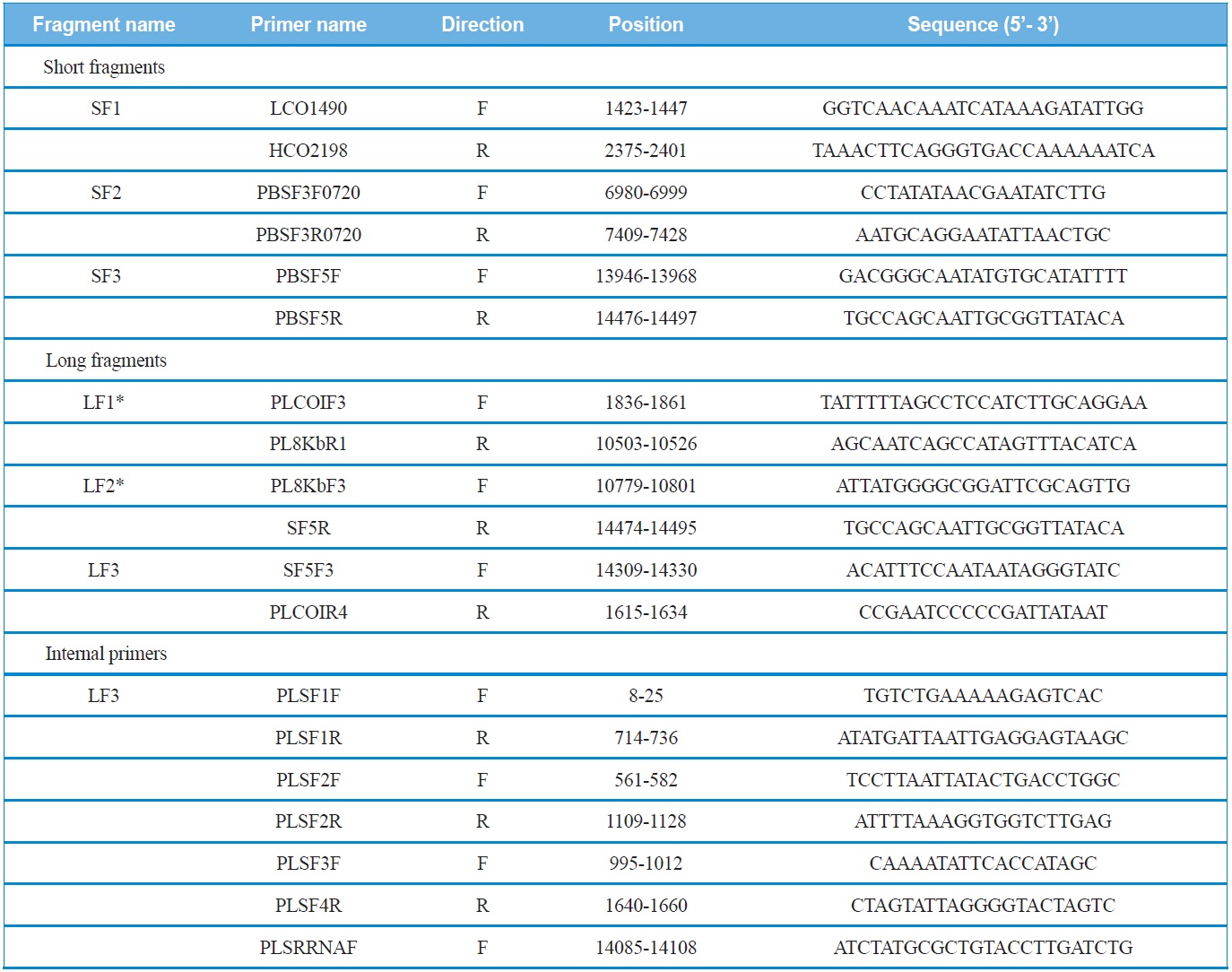
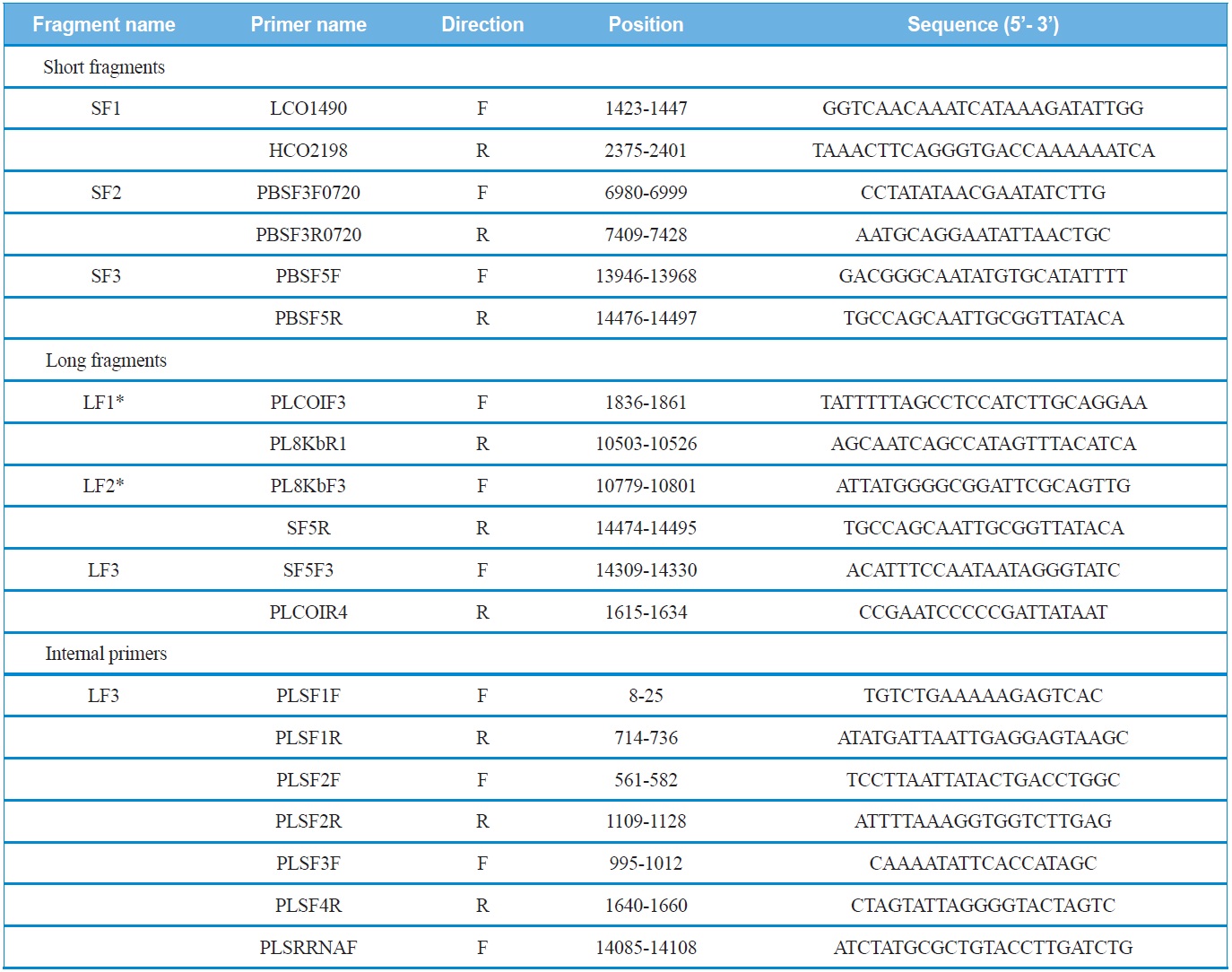
Three short fragments, corresponding each about 500~700 bp were amplified from three genes, such as COI (SF1), CytB (SF2), and srRNA (SF3) (Fig. 1). Primers for these
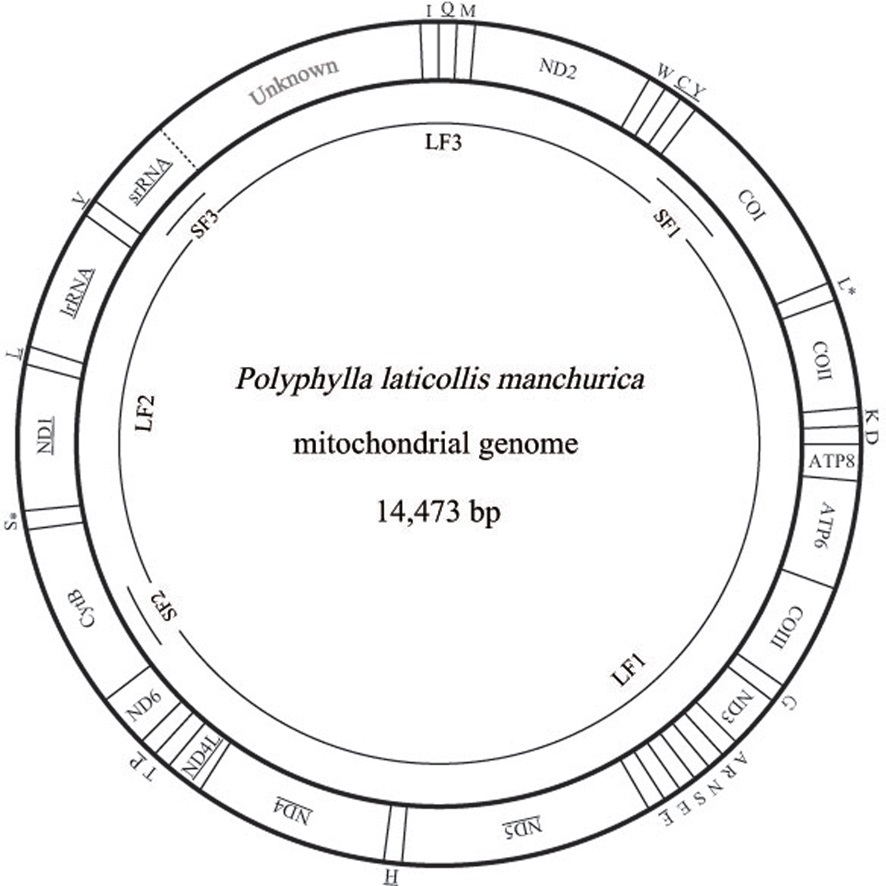
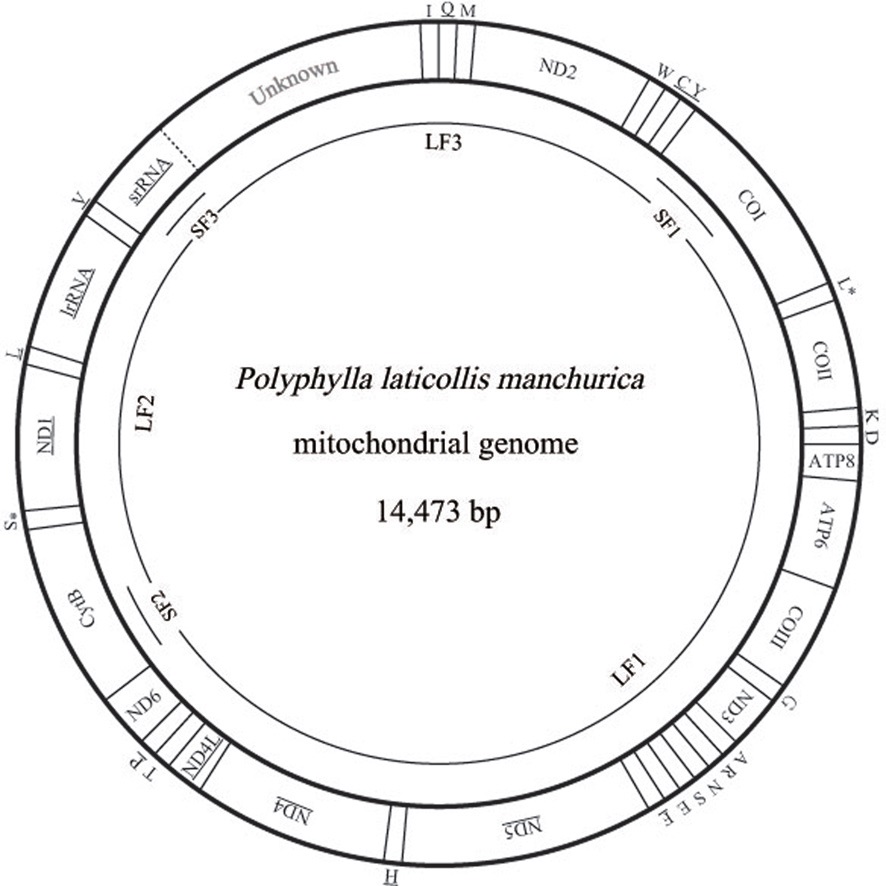
[Fig. 1.] Circular map of the mitochondrial genomes of Polyphylla laticollis manchurica. The abbreviations for the genes are as follows:
COI, COII, and COIII refer to the cytochrome oxidase subunits, CytB
refers to cytochrome B, ATP6 and ATP8 refer to subunits 6 and 8 of F0
ATPase, and ND1 ~ 6 refer to components of NADH dehydrogenase.
tRNAs are denoted as one-letter symbols consistent with the IUPACIUB
single letter amino acid codes. The one-letter symbols L, L*, S
and S* denote tRNALeu(CUN), tRNALeu(UUR), tRNASer(AGN), and
tRNASer(UCN), respectively. Gene names that are not underlined
indicate a clockwise transcriptional direction, whereas underlined
genes indicate a counter-clockwise transcriptional direction. The P. l. manchurica mitogenome was amplified each from three short (SF1,
SF2, and SF3) and three long (LF1, LF2, and LF3) overlapping
fragments, shown as single lines within a circle. The unknown region
that possibly contains partial srRNA and the A+T-rich region is shaded.
short fragments were designed via the alignment of several coleopteran mitogenomes sequenced in their entirety. These short fragments were amplified with AccuPower® PCR PreMix (Bioneer, Korea) using an initial denaturation at 94℃ for 4 min, followed by 35 cycles of 30 s denaturation at 94℃, 40 sec annealing at 50-55℃, and a 60 sec extension at 72℃. The final extension step was continued for 8 min.
Using the sequence information obtained from the short fragments, three primer sets specific to each species were designed to amplify three long fragments (LF1 ~ LF3) which overlapped with the short fragments (SF1 ~ SF3). PCR cycles were as follows: denaturation for 2 min at 96℃, followed by 30 cycles of 10 sec at 98℃ and 15 min at 58-65℃, and a final 10 min extension at 72℃. In order to sequence the long fragments, both primer walking and shotgun approaches were used, because the success of sequencing varied depending on fragments. Nevertheless, both approaches were unsuccessful for LF3, and, thus, the partial LF3 that encompasses the 5’-end of the srRNA and the whole A+T-rich region was unfinished. We believe this may have happened because this region is exceptionally long, containing unexpectedly long repeat regions and a high A/T content, considering previous sequence results of other coleopteran species, such as P. brevitarsis and M. blanchardi (Kim et al., 2013a, 2013b). Primer information for each short fragment, long fragment, and internal primers for primer walking are provided (Table 1).
For the primer walking method, internal primers were directly used to complete the sequences of the long fragment subsequent to purification with OIAquick PCR Purification Kit (Qiagen, USA). For the shotgun approach the long PCR fragments were subjected to shearing into 1~5 kb fragments (Gene Machine, USA) and were cloned into the pUC118 vector (Takara Biomedical, Japan). Each resultant plasmid DNA was isolated using a Wizard Plus SV Minipreps DNA Purification System (Promega, USA). DNA sequencing was conducted using the ABI PRISM® BigDye® Terminator v3.1 Cycle Sequencing Kit and the ABI PRISM® 3100 Genetic Analyzer (PE Applied Biosystems, USA).
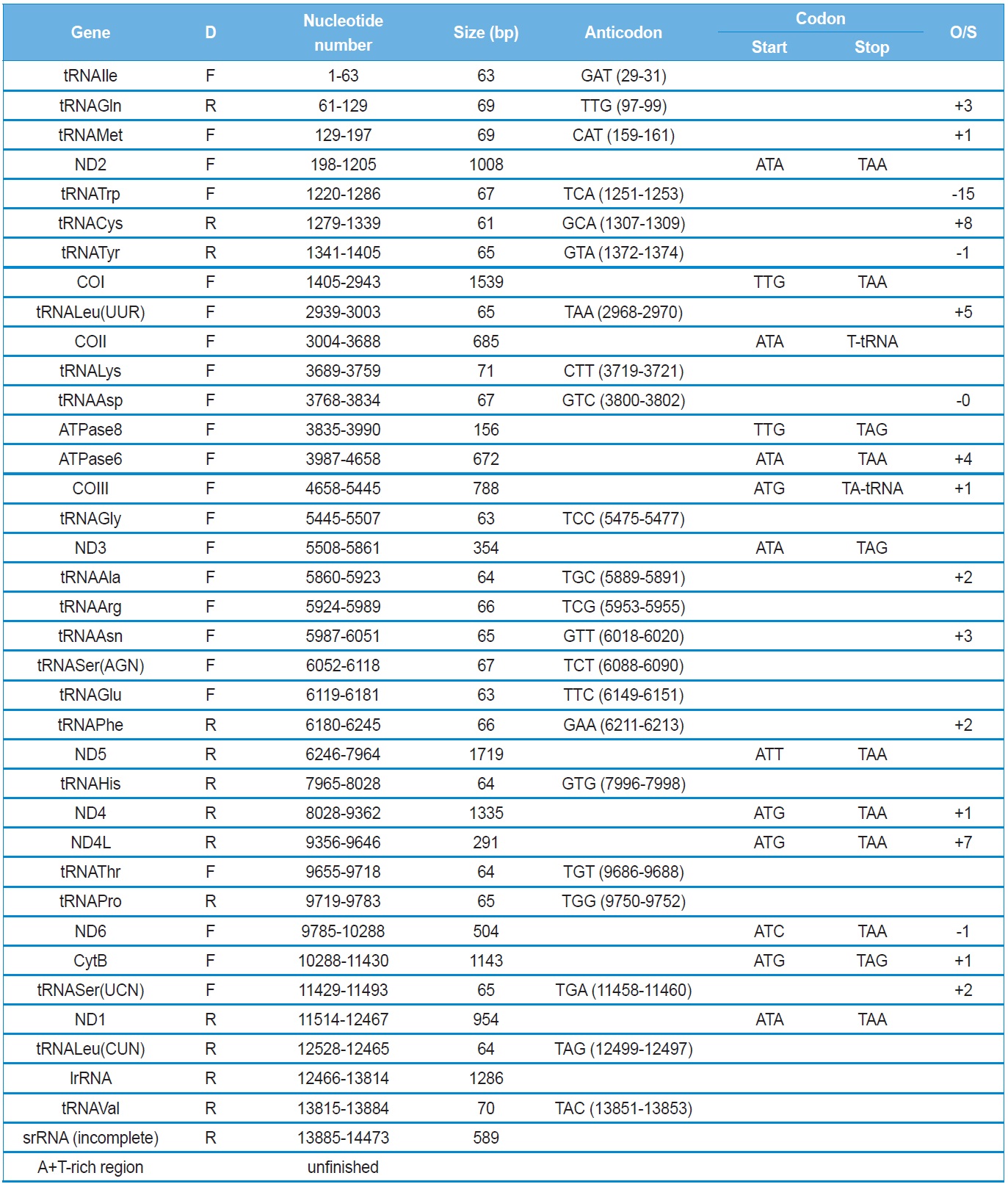
The boundary of individual thirteen mitochondrial protein-coding genes (PCGs) and individual two rRNAs were determined through the alignment of the homologous sequences of known full-length coleopteran mitochondrial genome sequences using the CLUSTAL X program (Thompson et al., 1997). The nucleotide sequences of the PCGs were translated on the basis of the invertebrate mtDNA genetic code (http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/index.cgi?chapter=cgencodes). The translated amino acid sequence was further utilized to identify and delimitate the 13 PCGs. The tRNA genes were identified by their predicted cloverleaf secondary structure and anticodon sequences using the tRNAscan-SE version 1.1 with the option of invertebrate codon predictors and a cove score cut off of 1 (Lowe and Eddy 1997). The tRNASer(AGN) and tRNAAsn were initially identified through alignment of the nucleotide sequences of the tRNA genes of known full-length coleopteran mtDNA sequences, and were further confirmed by hand-drawing their secondary cloverleaf structure by inspection of respective anticodon sequences. The sequence data determined in this study have been deposited to GenBank under the accession number KF544959.
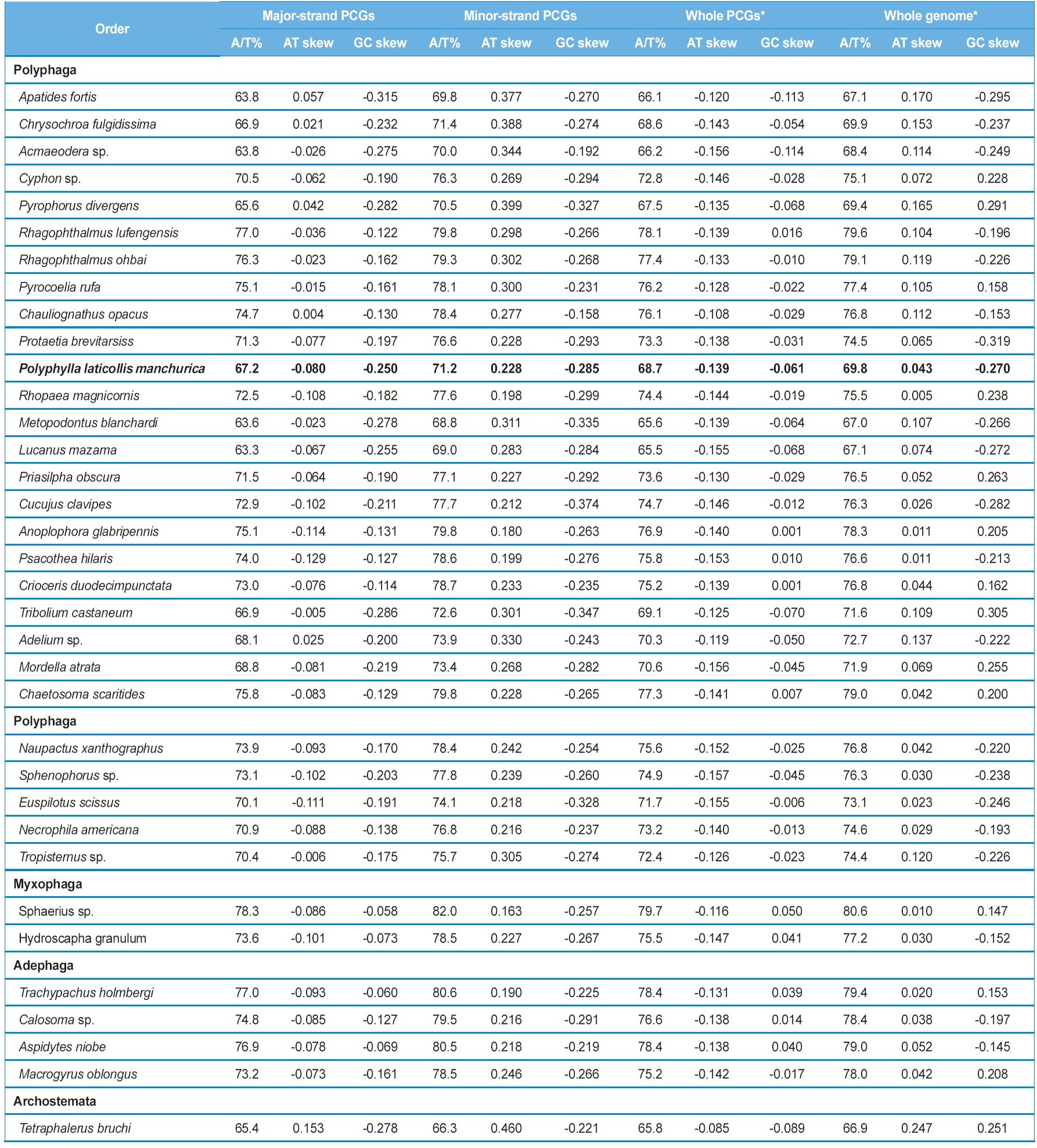
We compared 35 species of coleopteran insects including the currently sequenced P. l. manchurica. Nucleotide composition of each gene, whole genome, and each codon position of PCGs were calculated using DNASTAR (Burland, 2000). Frequencies of both codon and amino acid were calculated using SWAAP ver. 1.0.3 (http://www.bacteriamuseum.org/SWAAP/SwaapPage.htm). Gene overlap and intergenic-space sequences were handcounted. Nucleotide composition, termed “compositional skew” was calculated for the PCGs between two strands, and the whole genome with the EditSeq program included in the Lasergene software package (www.dnastar.com) using the following formula proposed by Perna and Kocher (1995): GC-skew = (G-C)/(G+C) and AT-skew = (A-T)/(A+T), where C, G, A, and T are the frequencies of the four bases.
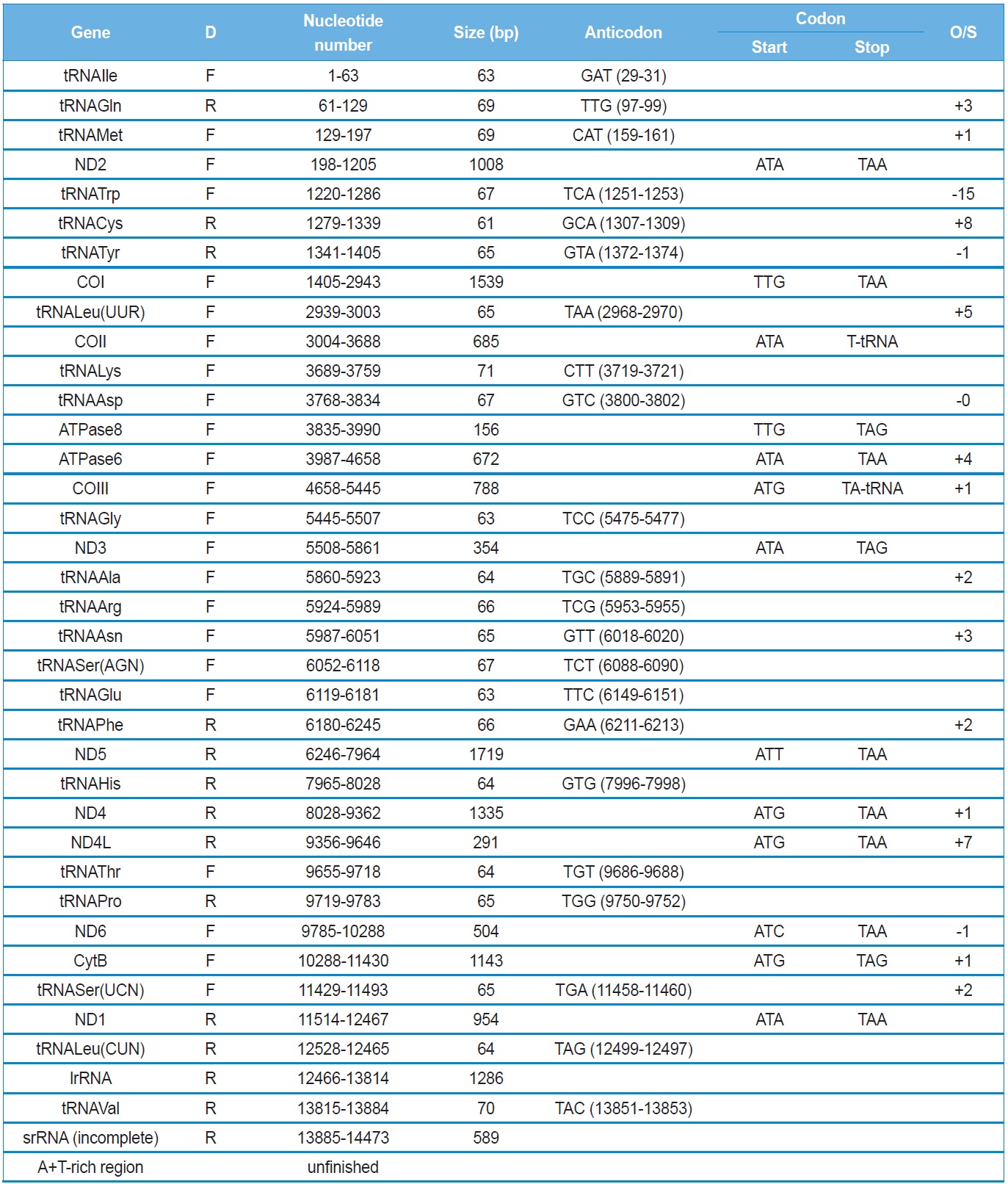
The Polyphylla laticollis manchurica mitogenome contains typical sets of genes, such as 13 PCGs, 22 tRNA genes, and 2 rRNA genes (Table 2). Typically, coleopteran insects contain one large non-coding region, named the A+T-rich region in insects, but we were unsuccessful in sequencing this region and the neighboring partial 5’-region of srRNA. In GenBank, a substantial number of incomplete mitogenome sequences lacking the A+T-rich region and neighboring sequences are registered and published as incomplete mitogenomes (Sheffield et al., 2008). Possible reasons for such failure may be the presence of secondary structural motifs and the excessive length of the A+T region. Several coleopteran species have been reported to have an extraordinary long A+T-rich region and an extra non-coding sequences neighboring to the region. For example, the seven-spotted lady beetle has been reported to have a 4,469 bp-long A+T-rich region which is composed of a 2,214 bp of non-repeat region and a 2,255 bp repeat region (Kim et al., 2012). Furthermore, the whiter-spotted flower chafer, P. brevitarsis, has a 5,646-bp long A+T-rich region which is composed of a 1,804 bp non-repeat region and a 3,841 bp repeat region (Kim et al., 2013a). In addition, insect mitogenomes infrequently contain substantially longer intergenic spacer sequences which are composed of tandem repeat unit located neighboring to the A+T-rich region. For example, the Korean firefly, Pyrocoelia rufa, has a 1724-bp long intergenic spacer (Bae et al., 2004). More extreme case is the two-spotted stag beetle,
which contains a 4,051-bp long, large non-coding sequence located between tRNAIle and tRNAGln, along with a 3,100-bp long A+T-rich region, resulting in a genome size of 21,628 bp, which is at least 5 kb larger than typical animal mitogenomes (Kim et al., 2013b). Considering these, it would be a reasonable speculation that the unfinished region is unusually large. The secondary structural motifs in the P. l. manchurica mitogenome might be another source that prevented the polymerases correctly passing across this region, resulting in casual amplification and making subsequent sequencing impossible (Carapelli et al., 2006). Several coleopteran mitogenomes which have extraordinary long A+T-rich region also have several long repeat sequences (Kim et al., 2012; 2013a, 2013b). These repeat sequences can easily form stem-andloop structures (Zhang and Hewitt, 1997; Kim et al., 2007), and thus, the unfinished A+T-rich region, along with the neighboring region of the P. l. manchurica mitogenome is highly likely to have such motifs. Nevertheless, this failure was not fully understood, considering our previous success in sequencing this region from several coleopteran insects (Kim et al., 2012, 2013a, 2013b).
The P. l. manchurica mitogenome has an identical gene order that has been found in the majority of insect species (Boore, 1998). Among 35 complete or nearly complete coleopteran mitogenomes (Table 3), only Tribolium castaneum differs from the most common type, by the movement of the tRNAGlu to a position 3’-downstream of tRNAPhe, thus resulting in an order of tRNAPhe and tRNAGlu, rather than the order tRNAGlu and tRNAPhe (Friedrich and Muquim 2003).
The P. l. manchurica mitochondrial genes harbor a total of 47 bp of intergenic spacer sequences, which are spread over six regions, ranging in size from 1 to 20 bp, with the longest being located between tRNASer(UCN) and ND1 (Table 2). Similar sized intergenic spacer sequences are found in the majority of sequenced coleopteran insects (16 ~ 22 bp), except for those of Rhagophthalmus lufengensis and R. ohbai, which are only 5-bp long (Li et al., 2007). Previously, it has been shown that the region contains a 5 bp-long motif sequence (TAGTA) in the coleopteran insects (Sheffield et al., 2008). Our additional analysis, including all available coleopteran species and P. l. manchurica, consistently revealed a motif sequence in the region (Fig. 2). This 5-bp consensus sequence was suggested to be the possible binding site for mtTERM, the transcription termination peptide, with the consideration that the intergenic spacer sequence is detected at the end site of the major-strand coding region in the circular mtDNA (Taanman, 1999).
Next longer intergenic spacer sequence is located between ND2 and tRNATrp as 14 bp (Table 2). A search on this region from other coleopteran insects has shown fluctuating length: only a few spacer sequences in most species (e.g., 4 bp in Pyrophorus divergens ; Arnoldi et al., 2007), abuttal between the two genes (e.g., Psacothea hilaris ; Kim et al., 2009), and overlapping between the two genes (2 bp in Tribolium castaneum ; Friedrich and Muqim, 2003). An exceptional case is that of P. rufa, which has a 1,724-bp long tandem repeat unit between the two genes (Bae et al., 2004). The P. l. manchurica mitochondrial genes overlap in a total of 33 bp at 12 locations, with the longest overlap measuring 8 bp, and located between tRNATrp and tRNACys. Similarly-sized overlapping sequences are also detected in several coleopteran insects (e.g., P. brevitarsis; Kim et al., 2013a; Metopodontus blanchardi ; Kim et al., 2013b), but abuttal (P. rufa ; Bae et al., 2004) and overlapping regions (8 bp in Pyrophorus divergens ; Arnoldi et al., 2007) are also found between the two tRNAs.
The P. l. manchurica mitogenomes harbors 3,704 codons, excluding termination codons, and this number is identical to those of Naupactus xanthographus (Song et al., 2010) and most similar to those of Sphenophorus sp. (3,705 codons; Song et al., 2010) and Tetraphalerus bruch (3705 codons; Sheffield et al., 2008).
All PCGs, with the exception of COI and ATPase8 of the P. l. manchurica mitogenomes have the typical ATN codon (Table 2). The initiation site for the COI gene and the precedent tRNATyr does not harbor the typical start codon, except for the infrequent, but typical invertebrate start codon TTG that is located at the beginning region of the COI, overlapping one nucleotide with the 5’-end of the tRNATyr (Fig. 3). Thus, we designated the TTA as the COI start codon. Only a few other sequenced coleopteran insects have a typical ATN start codon, but these are all located inside neighboring tRNATyr (Fig. 3). Thus, Sheffield et al. (2008) previously suggested that start codon for COI genes should be chosen to minimize
intergenic spacer sequences and gene overlaps. In this regard, they proposed asparagines (AAT or AAC) as the start codon for COI gene, because those are the first non-overlapping inframe codons, and found at the corresponding position in all sequenced Polyphaga in Coleoptera (Fig. 3; Sheffield et al., 2008). Furthermore, they hypothesized that asparagines may function as a molecular synapomorphy for Polyphaga (Sheffield et al., 2008). However, our newly sequenced P. l. manchurica, which belongs to Polyphaga, instead have AAG (Lysine) at the corresponding position and have the typical TTG precedent to AAG. Thus, our P. l. manchurica is the only example that does not follow the unanimous COI start codon in Polyphaga. The P. l. manchurica ATPase8 also has the TTG start codon (Fig. 4). Previously, the TTG codon was found in ND1 gene for coleopteran P. rufa (Bae et al., 2004) and dipteran Anopheles quadrimaculatus (Mitchell et al., 1993), and COI gene for lepidopteran Caligula boisduvalii (Hong et al., 2008), but has never been found for the start codon for ATPase8 in Coleoptera (data not shown).
Eleven of the 13 PCGs have a complete termination codon of TAA or TAG, but the COII and COIII genes harbor the incomplete termination codon, T (Table 2). The most common interpretation of this phenomenon is that TAA termini are created via post-transcriptional polyadenylation (Ojala et al., 1981).
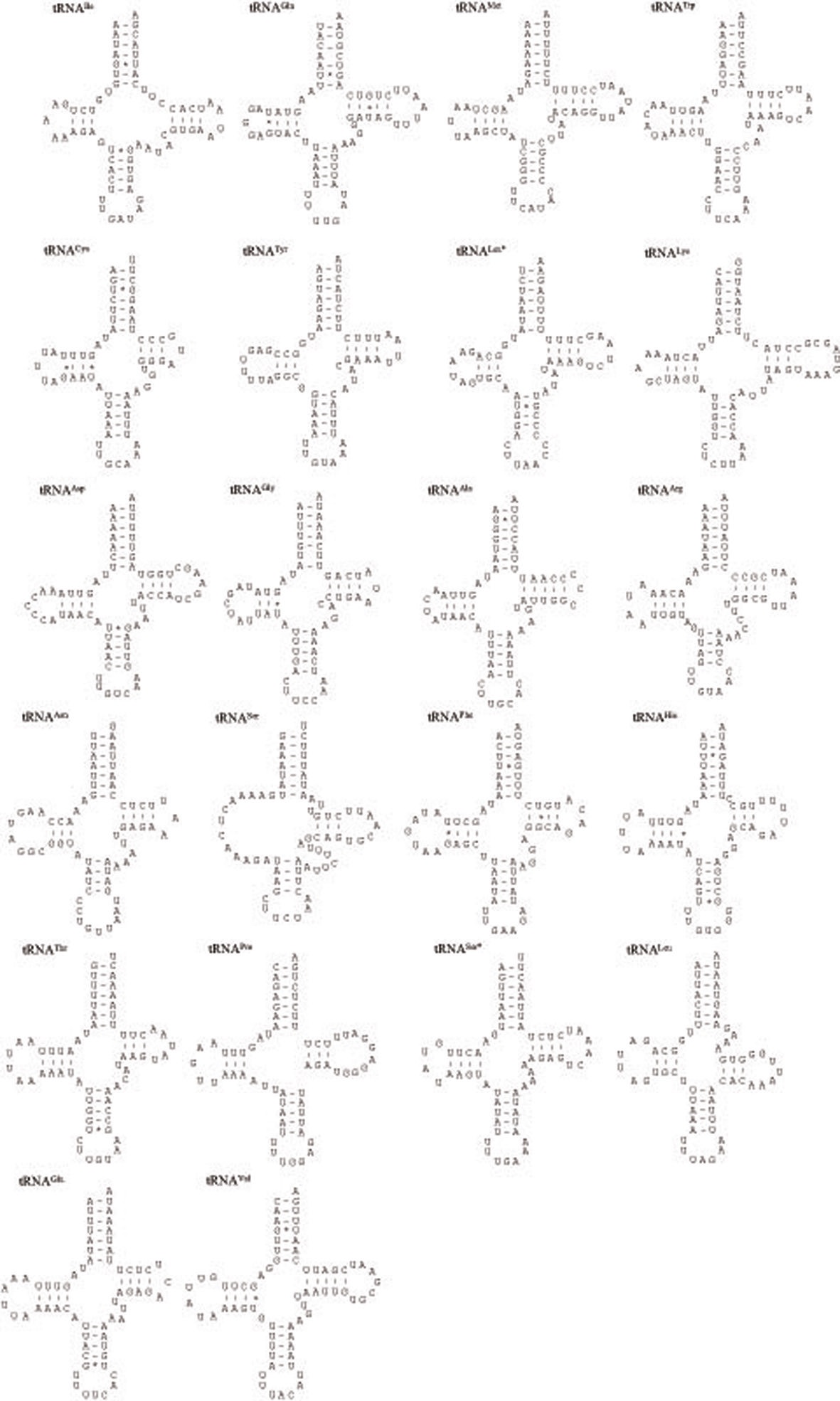
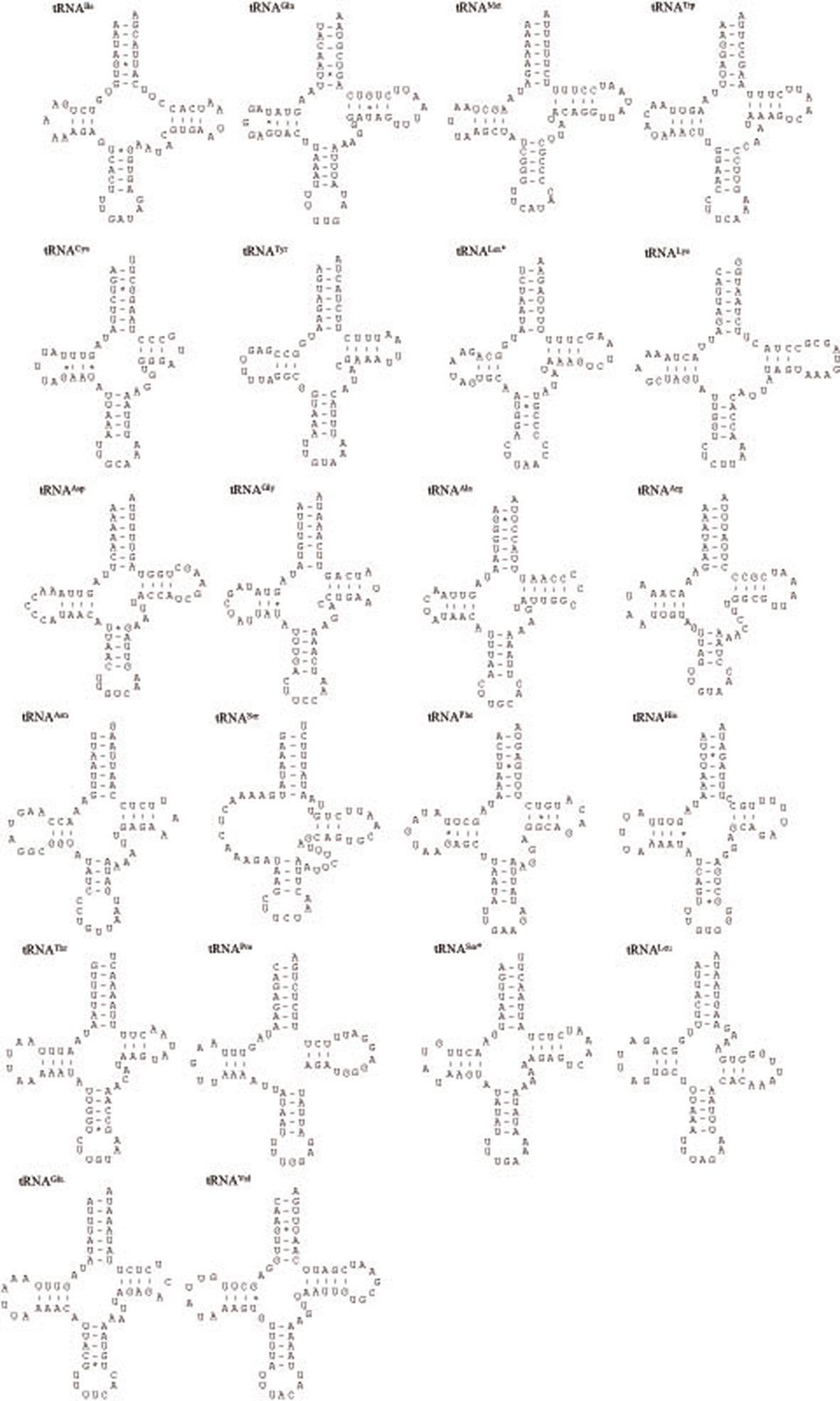
The P. l. manchurica mitogenomes harbors 22 tRNA genes that are interspersed thorough genomes (Fig. 5). Except for tRNASer(AGN), the dihydrouridine (DHU) arm of which forms a simple loop, all tRNAs were folded into the typical clover-leaf structure. The aberrant tRNASer(AGN) has been reported in many
metazoan species, including insects (Wolstenholme 1992; Garey and Wolstenholme, 1989). For the proper function of a tRNA the DHU arm, which is involved in tertiary interaction requires proper folding (Rich and RajBhandary, 1976). The nuclear magnetic resonance analysis from nematodes has shown that the aberrant tRNASer(AGN) also functions in a similar way to that of usual tRNAs by structural adjustment to fit in the ribosome (Ohtsuki et al., 2002).
The size of tRNAs ranged from 61 (tRNACys) to 71 bp (tRNALys) in P. l. manchurica (Table 2). Interestingly, tRNALys is often slightly larger than tRNAs in many other coleopteran mitogenomes. In fact, 29 among 35 coleopteran species analyzed in this study ranked the tRNALys as the longest tRNA at 71 bp (data not shown). In the varying tRNA size, the size of aminocyle stem as 7 bp, anticodon loops as 7 bp, and anticodon stem as 5 bp were all well conserved in all P. l manchurica tRNAs. Most of size variation among tRNAs stemmed from the length variation in DHU and TΨC arms, within which loop sizes (3 ~ 13 bp) are more variable than stem sizes (3 ~ 7 bp). With the numerous number of G-U base pairs, which form a weak bond in the tRNAs the P. l manchurica tRNAs contained 11 mismatches (three U-C, and each two A-C, U-U, G-A, and A-A mismatches) (Fig. 5). As with all other insect mitogenome sequences, two rRNA genes were detected in P. l manchurica. The lrRNA is located between tRNALeu(CUN) and tRNAVal and the srRNA is located between tRNAVal and the presumable A+T-rich region, respectively (Fig. 1).
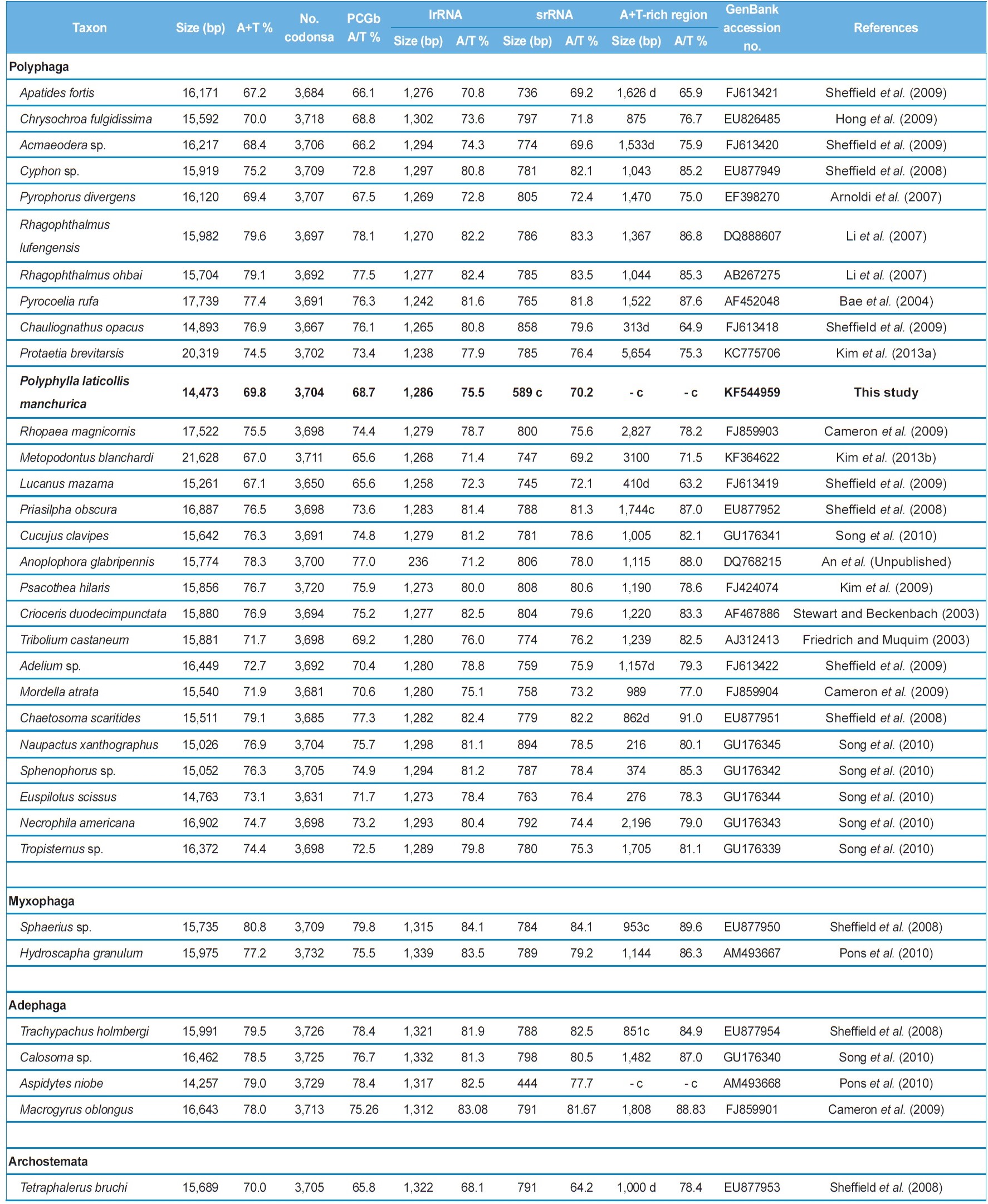
The nucleotide composition of the mitogenome of P. l. manchurica is also biased toward A/T content at 69.9% (Table 3). This value is well within the range found in the sequenced
[Fig. 5.] Predicted secondary clover-leaf structures for the 22 tRNA genes of Polyphylla laticollis manchurica. The tRNAs are labeled with
the abbreviations of their corresponding amino acids. The one-letter symbols L, L*, S and S* denote tRNALeu(CUN), tRNALeu(UUR),
tRNASer(AGN), and tRNASer(UCN), respectively. Arms of tRNAs (clockwise from top) are the amino acid acceptor (AA) arm, TΨC (T) arm,
the anticodon (AC) arm, and the dihydrouridine (DHU) arm. Nucleotide sequences from 5’ to 3’ are indicated from the left side of the amino
acid stem. Dashes (-) indicate Watson-Crick base-pairing, and centered asterisks (*) indicate G-U base-pairing.
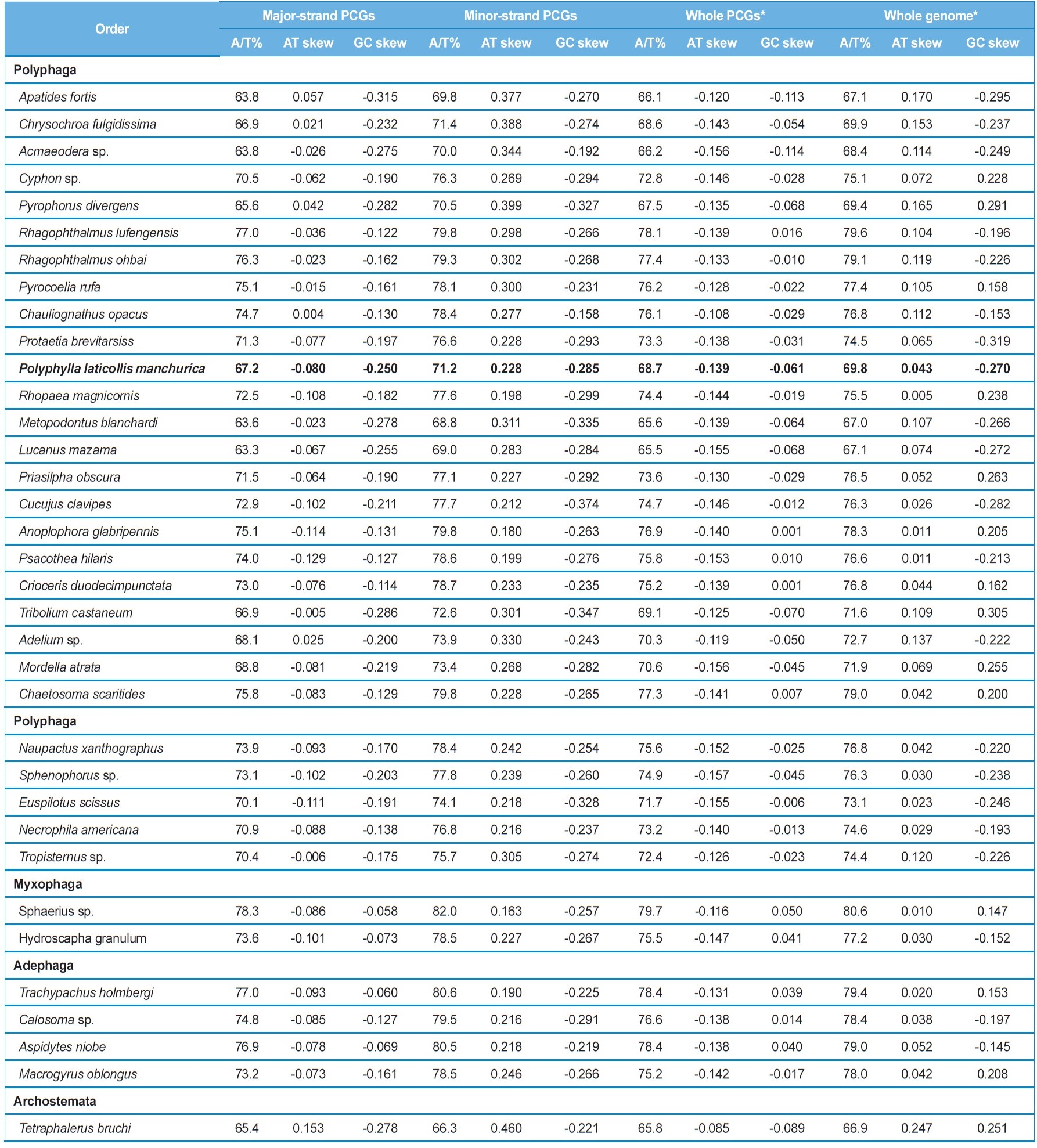
coleopteran insects, where these values range from 67.2% in Apatides fortis to 80.8% in Sphaerius sp. (Table 3). To evaluate the degree of the base bias, the AT-skew and GC-skew each in the whole genome and whole PCGs from the major strand and each major- and minor-strand PCGs was measured from coleopteran insects, including P. l. manchurica (Table 4). Overall, whole genomes of all coleopteran species including the P. l. manchurica are obviously A- and C-skewed, whereas whole PCGs are T- and C-skewed, indicating that Ts are clearly more favored over As in the PCGs, and the evolutionary pattern of PCGs differ from the remaining genes. In the majority of coleopteran species, including P. l. manchurica, the major-strand, in which nine PCGs (ND2,
ND3, ND6, COI, COII, COIII, ATPase6, ATPase8, and CytB) are encoded is slightly T-skewed (AT skew = -0.129 ~ 0.153), whereas the minor-strand, in which four PCGs (ND1, ND4, ND4L, and ND5) are encoded is obviously A-skewed (AT skew = 0.163 ~ 0.460), although both strands are C-skewed (Table 4). Thus, the two strands are sharply distinct in A/T-skewness, indicating that mutational pressures that favor Ts or As are starkly different between the two strands. It has been suggested that the lagging strand, equivalent to the major strand, should be more prone to chemical conversion of As to Gs and Cs to Ts, by the mechanism called deamination than the leading strand, and this may have resulted in the enriched Ts and Gs in the lagging strand, and As and Cs in the leading strand (Reyes et al., 1998). Nevertheless, current coleopteran mitogenomes pretty strongly display C-skewness rather than G-skewness in the major strand (Table 4). Thus, the strand-based inequality has yet to be clearly understood.
The genome-wise A/T bias is also reflected in the codon usage of P. l. manchurica mitogenomes (Table 5). The codons TTA (Leu), ATT (Ile), TTT (Phe), and ATA (Met) are the four most frequently used codons in the P. l. manchurica PCGs, accounting for 28.22% and on average these four codons accounted for 31.98% in Coleoptera (Table 5). Considering 60 codons are available, excluding each two start and stop codons (ATA, ATG, TAA, and TAG) the overuse of these four codons is obvious. These four codons are all comprised of A or T nucleotides, thus indicating the biased usage of A and T nucleotides in the coleopteran insects including P. l. manchurica PCGs.
The analysis of the base composition at each codon position of the concatenated 13 PCGs of P. l. manchurica showed 68.6% of the A/T content in the third codon position. This value is higher than that of the first (66.4%) and second (66.2%) codon positions, and on average, the A/T content in the third codon position was 74.9%, ranging from 65.4% to 82.1% in Coleoptera (Table 6). In a comparison among species-diverse insect orders, Hymenoptera is the highest on average at 93.1% (Hong et al., 2008), Lepidoptera is next at 92.1% (Kim et al., 2010), and Diptera rank third at 92.0% (Cameron et al., 2009), indicating that the Coleoptera is the least A/T-biased. Probably, this aspect has implications for phylogenetic analysis because of the different degrees of A/T bias, resulting in compositional heterogeneity among insect orders as a major source of systematic bias in phylogeny (Jermiin et al., 2004; Sheffield et al., 2009).