Direct methanol fuel cells (DMFCs) are considered promising candidates for mobile and transport applications due to their high energy density, zero emissions, and relatively low operating temperature [1]. Moreover, platinum (Pt) is an excellent catalyst and one of the best electrode materials for DMFCs [2-4]. However, the high cost, low electrocatalytic activity and stability of common Pt catalyst inhibit their broad application for DMFCs [3]. It is well known that the electrocatalytic performance of a fuel cell greatly depends on the composition, shape, size, and dispersion of the catalyst nanoparticles (NPs). The materials supporting catalyst play an important role in controlling these properties [5]. However, during fuel-cell operation, a gradual oxidation of the carbon support leads to detachment of Pt NPs from the carbon support, allowing their agglomeration, which can reach an unacceptable level [6].
In the past few years, graphene has emerged as the ideal material for a variety of different energy applications including fuel cells, Li-ion batteries, supercapacitors, and solar cells. This is because of its large thermal conductivity, high surface area, high mechanical stability, and excellent electrical conductivity [7]. Chemical modification of the inert surface of graphene sheets is necessary for the uniform dispersion of Pt NPs. Currently, chemical modification is achieved by methods including acid oxidation, ionic liquid [8], and conductive polymer [9].
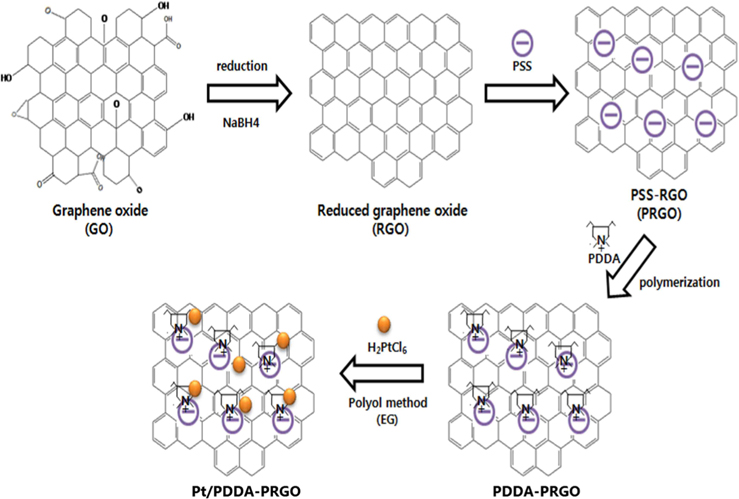
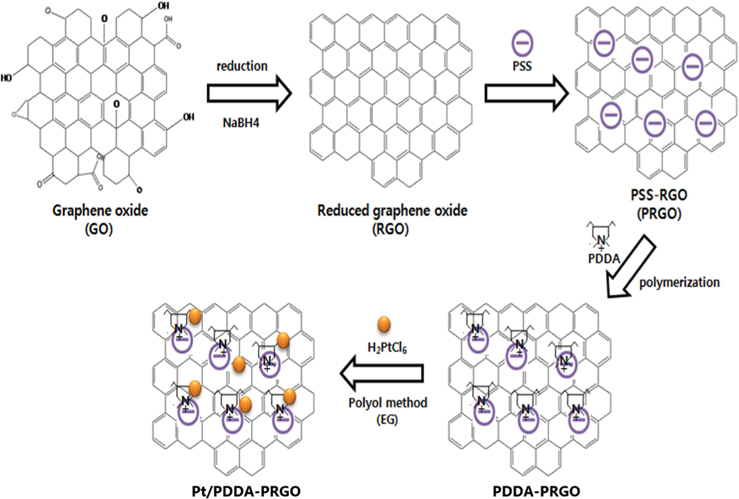
Recently, we developed a facile and efficient route to decorate Pt NPs onto reduced graphene oxide (RGO) functionalized with poly(diallyldimethyl ammonium chloride) (PDDA), as acationic polymer. In this work, we investigated the synergistic effect of a poly(sodium 4-styrenesulfonate) (PSS)-functionalized graphene support and PDDA polymer for obtaining better electrocatalytic performance for DMFCs. These results demonstrated that, with high electronic conductivity and easier charge transfer, Pt/PDDA- PSS-functionalized RGO (PRGO) exhibited higher activity and stability than did Pt-decorated PDDA-RGO (Pt/PDDA-RGO), and Pt-decorated RGO (Pt/RGO).
PSS (MW = 70,000) and PDDA (20 wt% in water) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Graphite (99.9%) was purchased from Bay Carbon Inc. (Bay City, MI, USA) and H2PtCl6 6H2O was purchased from Sigma-Aldrich. Ethanol (99.8%), methanol (99.8%), and ethylene glycol (EG, 99.0%) were purchased from DAE JUNG Company and used without further purification. Distilled water was used in all preparation experiments.
Graphene oxide (GO) was synthesized from graphite powders using a modified Hummer’s method [10]. Briefly, graphite powders were first oxidized by sulfuric acid (H2SO4), sodium nitrate (NaNO3), potassium permanganate (KMnO4) for the acid treatment. Deionized (DI) water and hydrogen peroxide (H2O2) were put into the mixture with strong stirring at 90℃ for 4 h. Finally, DI water and hydrogen chloride (HCl) was put into the mixture. After removal of residual salts and acid, the resultant dispersion of graphite oxide was mixed with DI water and the sodium borohydride (NaBH4) used as reducing agent slowly added at 90℃, in order to get the RGO. To functionalize RGO with PSS, 0.2 g RGO was added to a flask with anionic polyelectrolyte PSS (0.15 mg mL−1) after dispersion by ultrasonication and stirred at 50℃ for 12 h. After that, the product was separated by centrifugation, washed, and freeze-dried overnight. The as-synthesized products were labeled PSS-functionalized RGO (PRGO) [11].
PRGO (0.2 g) was loaded in a 250 mL flask and PDDA solution (0.5 wt%, 100 mL) was added, yielding an inhomogeneous black dispersion. The mixture was heated in a reflux process at 90℃ for 5 h. The final product was collected by filtration and freeze-dried overnight. It was labeled PDDA-PRGO. For comparison, the PDDA coating procedure was also applied to RGO. The final product was labeled PDDA-RGO.
Pt/PDDA-PRGO with 20wt% Pt loading was prepared by a polyol process. PDDA-PRGO was vigorously mixed with H2PtCl6·6H2O in EG, after which the pH was adjusted (10-11) by adding NaOH (1.0 M). The solution was ultrasonicated for 20 min and exposed to N2 bubbling for 20 min, and then stirred for 4 h at 120℃. After reaction, the resulting black solid product was isolated by centrifugation, washed with distilled water and ethanol to remove the ions possibly remaining in the final products, and then freeze-dried overnight. For comparison, Pt/PDDA-RGO, and Pt/RGO were also synthesized by the above procedure, substituting PDDA-PRGO with PDDA-RGO and RGO, respectively (Fig. 1).
The infrared (IR) spectrum of the Pt/PDDA-PRGO was recorded by Fourier transform infrared spectra (FT-IR) (Spectrum GX, Thermo, Waltham, MA, USA). The particle size and morphology of the graphene, and the Pt/PDDA-PRGO or Pt/PDDA-RGO catalyst, were analyzed using transmission electron microscopy (TEM, H-7600, HITACHI, Tokyo, Japan). For the TEM characterization, the catalyst was ultrasonically dispersed in ethanol solution for 30 min to obtain a uniform ink. Subsequently, the ink was dropped onto copper grids covered with carbon film and dried in the air. Electrochemical experiments were performed on an IVIUMSTAT (Ivium Technologies, Eindhoven, the Netherlands). A three-electrode cell, consisting of a glassy carbon working-electrode, a Pt-wire counter-electrode, and an Ag/AgCl reference-electrode, was used. All potentials of the electrochemical measurement were converted to Ag/AgCl potential. The working electrode was prepared on a glassy carbon electrode by the following method. A suspension containing 5 mg (as catalysts) and 0.1 mL of 5wt% Nafion solution (Aldrich) in DI water (0.5 mL) was ultrasonically dispersed for 30 min, and then 30μL of the slurry was pipette onto the polished surface of the glassy carbon electrode. The solvent was completely evaporated in a vacuum oven over 30 min. The electrolyte was a 1M H2SO4 solution, or a mixture of 2M H2SO4 and 1M CH3OH solution by cyclic voltammetry (CV) between −0.2 V and 1.0 V, at room temperature.
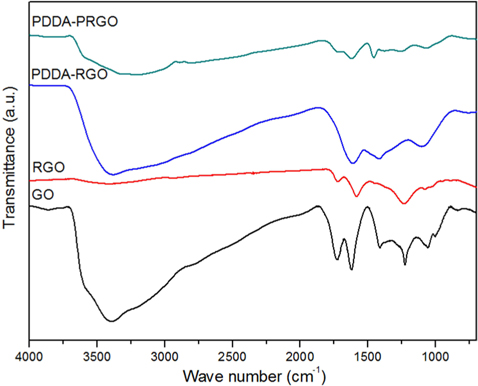
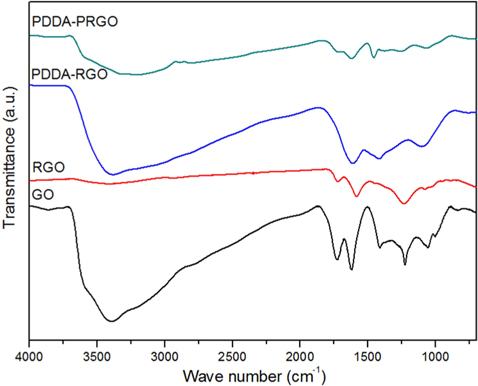
Fig. 2 shows the FT-IR patterns of PDDA-PRGO, PDDA-RGO, and GO recorded in the range of 4000-500 cm–1. The characteristic features of GO were the absorption bands corresponding to O–H stretching at 3405 cm−1, broadly, C=O stretching vibration of carboxyl at 1729 cm−1, C–OH stretching at 1222 cm–1, and C–O vibration at 1048 cm−1 [12]. Meanwhile, in the spectra of PDDA-PRGO, the characteristic peak of O-H stretching at 3405 cm−1 and C=O stretching vibration of carboxyl at 1729 cm−1 disappeared. However, O–H deformation at 1461 cm−1, and C–O vibration at 1067 cm−1 were obvious. These were the result of interaction between graphene and the remaining moisture. The C–C skeleton vibration of the carbon ring in graphene was at 1615 and 1646 cm−1. In addition, the absorption bands at 1467 cm−1 were associated with the C–N stretching of the PDDA [3]. From the FT-IR results, it was confirmed that the PDDA successfully coated the surface of the graphene.
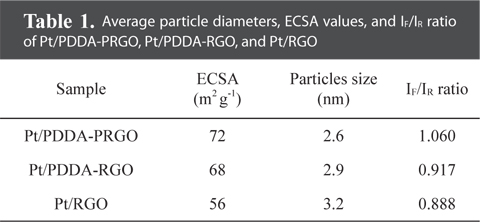
The particle size and dispersion of the Pt NPs decorated on PDDA-PRGO were studied using TEM. Fig. 3 shows TEM images of Pt/RGO (Fig. 3a and b), Pt/PDDA-RGO (Fig. 3c and d) and Pt/PDDA-PRGO (Fig. 3e and f), respectively. From Fig. 3a and b, it can be seen that the synthesized Pt NPs are poorly dispersed on RGO, leading to low efficiency of the catalyst. However, a uniform size distribution of Pt NPs on the surface of PDDA-RGO and PDDA-PRGO, with good dispersion, was observed in other TEM images (Fig. 3c-f). This could be ascribed to the presence of the PDDA molecules as stabilizer. The driving force for the stabilization of Pt NPs with PDDA is the coordination interaction between isolated electron pairs on the nitrogen atom of PDDA molecules, and the unoccupied d-orbit of Pt [13]. Additionally, the presence of PSS on RGO could serve as the active site for coating PDDA with a positive charge. Therefore, Pt/PDDA-PRGO has the best surface dispersion of Pt NPs. The statistical average particle sizes of Pt NPs supported on PDDA-PRGO, PDDA-RGO and RGO are 2.6, 2.9, and 3.2 nm, respectively. This is mainly because the PDDA-PRGO composite exhibited the synergistic effect of both PSS and PDDA polymers on RGO.
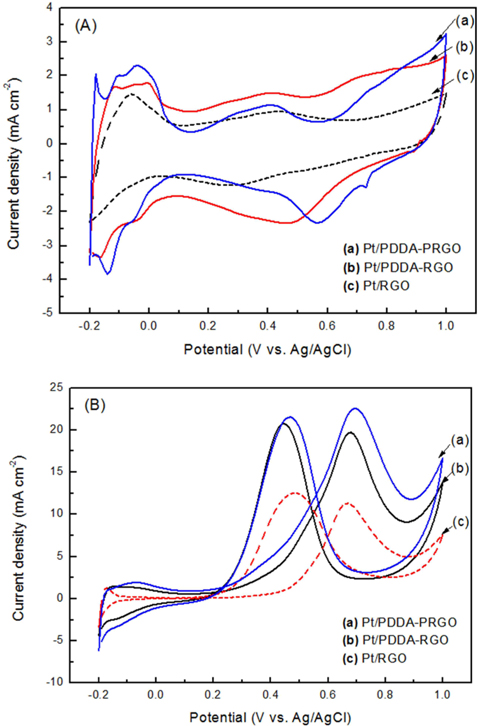
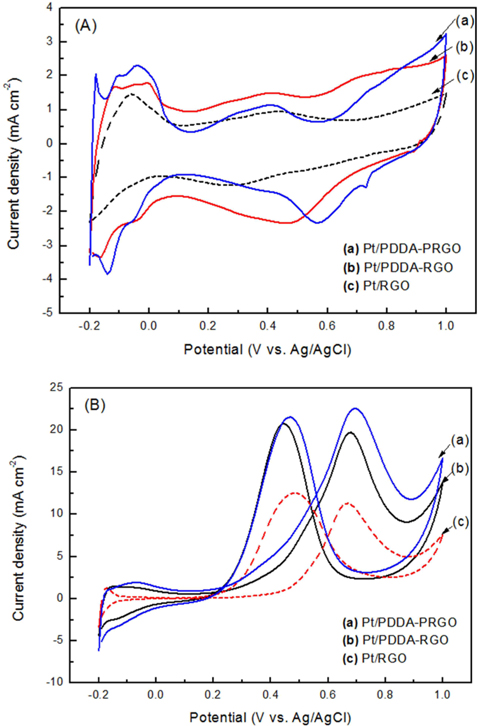
The CV of (a) Pt/RGO, (b) Pt/PDDA-RGO and (c) Pt/PDDA-PRGO catalysts in a de-aerated solution of 1.0 M H2SO4 is shown in Fig. 4a. The potential scan started at −0.2 V and ended at 1.0 V, and then went back from 1.0 V to −0.2 V. Well-defined hydrogen desorption/adsorption peaks in the potential range of −0.2 to 0.05 V vs. Ag/AgCl and irreversible pre-oxidation/reduction peaks in the potential range of 0.5 to 1.0V vs. Ag/AgCl are observed. The electrochemical surface area (ECSA) was determined by considering the CV area [14]. The ECSA was calculated by using the following eq [15]:
ECSA = QH / ([Pt] X 0.21)
QH is the charge density for hydrogen desorption (mC cm−2); [Pt] represents the Pt NP loading in the electrode (g cm−2); and 0.21 is the theoretical charge required to oxidize a monolayer of H2 on a flat Pt surface (mC cm−2). The ECSA evaluated from hydrogen adsorption/desorption peaks, was calculated to be 72, 68, and 56 m2 g−1 for Pt/PDDA-PRGO, Pt/PDDA-RGO, and Pt/RGO catalysts, respectively. The higher ECSA of Pt/PDDA-PRGO was most likely due to the smaller size and much better dispersion of the Pt NPs on PDDA-PRGO.
The CVs of methanol oxidation on different composites are shown in Fig. 4b, which were recorded in 1M H2SO4 + 2M CH3OH. Two peaks indicating methanol oxidation under anodic condition were clearly observed. The oxidation peak in the forward scan corresponded to the oxidation of freshly chemisorbed species coming from methanol adsorption; the reverse oxidation peak was primarily associated with removal of carbonaceous species not completely oxidized in the forward scan, rather than being caused by freshly chemisorbed species. The ratio of the forward-scan peak current (IF) to the reverse-scan peak (IR) could be applied to describe the electro-catalyst capability of the intermediate carbonaceous species accumulated on the electrode surface [15,16]. The IF/IR ratio became higher and higher, meaning better removal of CO gas poisoning from the electro-catalyst surface [16]. Table 1 shows the values of IF and IR, respectively. The IF/IR ratio decreased as the scan rate increased, indicating that the CO accumulation increases with increasing scan rate. The higher anodic current of Pt/PDDA-PRGO indicated that the electric conductivity of graphene was higher than the commercial carbon. All of the above data revealed that Pt/PDDA-PRGO exhibits enhanced catalytic activity for methanol oxidation.
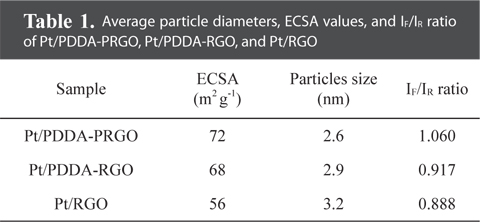
Average particle diameters, ECSA values, and IF/IR ratio of Pt/PDDA-PRGO, Pt/PDDA-RGO, and Pt/RGO
Herein, we present a controllable route to prepare Pt NPs on graphene oxide via PDDA-PRGO. The Pt NPs on the support PDDA-PRGO were synthesized by EG reduction. This is unique in that PDDA-PRGO exhibits excellent stabilization effect and sufficient protection against aggregation of Pt NPs. These Pt NPs, which were dispersed on PDDA-PRGO, were 2.6 nm in diameter and had a very narrow particle-size distribution. The ECSA of Pt/PDDA-PRGO was enhanced to 72 m2 g−1, compared with Pt/PDDA-RGO (68 m2 g−1) and Pt/RGO (56 m2 g−1). Moreover, compared with traditional Pt/PDDA-RGO and Pt/RGO catalysts, Pt/PDDA-PRGO exhibits great tolerance towards poisoning by CO. The high activity and good stability of Pt/PDDA-PRGO catalyst, attests to the excellent dispersion of the Pt NPs, and to the surface properties of GO. These results indicate that Pt/PDDA-PRGO is a promising catalyst for fuel cell applications.