개별 생물의 유전적 특성인 유전형 정보를 얻기 위한 개발된 기법들 중 현재 가장 많이 사용되고 있는 것은 차세대 염기서열결정을 통해 얻어진 서열을 분석하여 단일핵산염기다형현상 기반의 유전형 정보를 얻어내는GBS 방법이다. 현재 TASSEL은 GBS방법을 통해 얻어진 서열을 분석하여 시료의 유전형을 측정하기 위해 가장 많이 사용되고 있는 프로그램 중 하나이다. 그러나 TASSEL은 염기서열결정을 통해 얻어진 서열 중 일부만을 사용하는 한계가 존재한다. 우리는 이러한 한계를 극복하기 위한 효율성 개선에 대한 연구를 시작하였다. 효율성 개선을 위해 TASSEL에서 사용후 버려지는 서열의 퀄리티를 체크하여 에러율 0.1% 이하인 데이터를 확인 한 후 퀄리티가 에러율을 충족하는 부분의 서열들을 필터링 한다. 그리고 마지막으로 바코드와 제한 효소의 부분을 확인하여 길이에 따라 서열을 잘라내어 새로운 데이터 셋으로 생성하는 구조를 반복하는 알고리즘으로 구현 하였으며, 약 17% 이상의 SNP 탐지효율성 증가함을 확인 하였다. 본 논문에서는 이와 같이 유전형 연구에서 사용되지 않는 유전체 염기서열들을 사용하여 더 많은 숫자의 단일 염기 다형성을 탐지하는 방법과 구현된 프로그램을 제시한다.
Recently, the most popular technique to determine the Genotype, genetic features of individual organisms, is the GBS based on SNP from sequences determined by NGS. As analyzing the sequences by the GBS, TASSEL is the most used program to identify the genotypes. But, TASSEL has limitation that it uses only the partial sequences that is obtained by NGS. We tried to improve the efficiency in use of the sequences in order to solve the limitation. So, we constructed new data sets by quality checking, filtering the unused sequences with error rate below 0.1% and clipping the sequences considering the location of barcode and enzyme. As a result, approximately over 17% of the SNP detection efficiency was increased. In this paper, we suggest the method and the applied programs in order to detect more SNPs by using the disused sequences.
개별 생물이 가지고 있는 유전체의 특성을 밝혀낸 유전형(genotype) 정보는 집단유전체학 등 학문적인 측면뿐만 아니라 실용적인 측면인 유전자 표지자(genetic marker) 개발 등 다양한 분야에 활용될 수 있는 정보이다. 개체의 크고 작은 유전형의 차이들 중 대부분은 유전체 간 개별 염기서열의 차이인 단일 염기 다형성(SNP: Single Nucleotide Polymorphism)으로 SNP 정보중 일부는 특정 유전 형질과 직접적으로 연관이 되어 유전형 마커(Genotyping Marker) 개발을 통한 작물의 육종에 활용되거나 작물 특성 연구의 주요 요인 혹은 집단 내 개체간의 유전적인 연관 관계 연구하는데 활용된다. 따라서 SNP을 탐지하기 위한 다양한 기법들이 다양하게 연구되어 왔다[1,7,9].
최근 유전체 연구에 있어 차세대 염기서열 분석 기법(NGS: Next Generation Sequencing)이 연구되고 발달함에 따라 염기서열 분석의 비용과 시간이 크게 감소되었다. 이에 따라 NGS를 이용해 과거에는 긴 시간과 노력이 필요했던 유전체 전체의 염기서열 분석(WGS: Whole Genome Sequencing) 또는 재염기서열 분석(Resequencing)을 적은 자원으로도 효과적으로 진행할 수 있게 되었다. 뿐만 아니라 그 효율성으로 인해 유전체의 구조, 유전변이, 차별적인 유전자의 발현, 전사 조절에 관한 연구 등 다양한 부분에서 사용되어지고 있다. 이러한 흐름은 생물의 유전형 파악에도 활용되어 시퀀싱을 통해 효과적으로 유전형을 파악할 수 있는 (GBS: Genotyping By Sequencing)기법이 개발되었다. GBS 분석은 크게 두단계로 나뉘며 그 첫 번째 단계는 시료로부터 유전체 정보를 분리하여 NGS를 이용해 염기서열결정(Sequencing) 수행하는 실험단계이고, 두 번째는 분석파이프라인을 이용하여 염기서열을 분석하여 유전형 정보를 분석해 내는 단계이다.
다양한 분석 파이프라인 중 가장 많이 사용되어지고 있는 GBS 분석 파이프라인은 코넬대학교 Buckler lab 에서 개발한 TASSEL(Trait Analysis by aSSociation, Evolution and Linkage)[2,8]로 현재 가장 안정적이고 우수한 결과를 보여주고 있다. 그러나 TASSEL은 바코드 형식의 태그가 부착된 전체 염기서열 중에서 통상 낮은 오류값을 가지는 것으로 알려진 서열의 시작부분만을 분석에 사용하는 특징이 있다. 우리는 이 사용되지 않는 염기서열을 분석하여 낮은 오류값을 가지고 있음에도 분석되지 않는 서열부위가 있을 수 있음을 확인하였으며 이를 사용함으로서 SNP 탐지에 대한 효율을 개선 할 수 있는 방법을 개발했기에 본 논문에서 제시하고자 한다.
본 논문은 제1장에서 서론을 기술한다. 제2장에서는 관련 연구를 분석, GBS방법 및 TASSEL 에 대해 설명한다. 제3장에서는 프로그램 알고리즘과 구현 그리고 비교 및 검토를 하고, 마지막 제4장에서 결론을 내리고, 향후 연구방향에 대해 기술한다.
현재까지 SNP 기반 유전형 측정을 위한 다양한 방법들이 보고되어 왔다. 이들 중 NGS를 이용한 SNP 분석 방법으로는 제한 효소 기반의 RAD-seq (Restriction site Associated DNA sequencing)가 먼저 개발되었다. 대표적인 분석 프로그램으로는 Julian M. Catchen 등이 발표한 Stacks[3]이 있었으며 이를 이용하여 개체 및 집단에서 SNP를 식별하였다. 다만, RAD-seq 방법은 실험방법이 복잡할 뿐만 아니라 양질의 결과를 얻기 위해서는 많은 양의 유전체 염기서열결정을 해야 하기 때문에 상대적으로 효율이 낮다.
이러한 단점을 극복하기 위해 나온 방법이 GBS로 상대적으로 적은 양의 염기서열결정 만으로도 RAD-seq과 동일한 수준의 결과를 얻을 수 있다. GBS는 다양한 작물의 종과 개체들의 SNP 유전형을 탐지하기 위한 목적으로 만들어진 NGS 기술의 최신 방법 중 하나이다. 다른 유전형 분석 기술과는 달리, GBS는 저렴한 비용으로 높은 수준의 SNP 마커들을 참조 유전체에 맵핑 할 수 있다. GBS 분석의 첫 번째 단계는 반복적인 지역의 유전체 서열을 피하고 동시에 유전체의 주요 지역이 선택될 수 있도록 하기위해 유전체 분석을 통해 가장 효과적인 제한효소를 선택하는 것이다. 다음으로 유전체를 제한효소로 처리한 후 서열의 양쪽 모두가 제한효소로 단편들 모두를 시퀀싱한다.
이러한 방법은 유전체 전체를 분석하지 않고도 넓은 유전체 범위에 대해 일정한 부분을 높은 비율로 분석할 수 있게 됨으로서 비용 및 시간을 감소시킨다. GBS는 Reduced Representation Library (RRL), RAD-seq 등과 같이 제한 효소를 이용하는 기본원리는 동일하지만 제한 효소로 자른 후 사이즈 크기를 상관하지 않는 점에서 라이브러리 제작이 더 간단하다[5].
TASSEL은 코넬대학교 Buckler lab에서 개발한 GBS 등 유전체와 제한 효소 정보를 이용한 유전형 분석을 위한 자바 기반의 분석 프로그램으로 개체군과 양적 유전학 도구로서 유전형과 특성 연관을 평가하는 소프트웨어이다[4]. TASSEL은 Discovery와 Production의 2개의 커다란 파이프라인[2]으로 이루어져 있다. Discovery 파이프라인은 바코드와 제한 효소로 처리가 되어 FASTQ 형식의 서열 정보를 이용하여 일정한 길이의 유전체 조각인 Tag들을 추출하고 이를 참조 유전체에 맵핑을 시킨 후 맵핑이 완료된 데이터로 부터 SNP를 탐지하는 역할을 한다. Production 파이프라인은 FASTQ 형식의 유전체 파일과 Discovery를 통해 맵핑된 데이터를 가지고 최종적으로 다수의 시료에 대한 Hatmap 데이터 포맷의 유전형 정보를 생성한다.
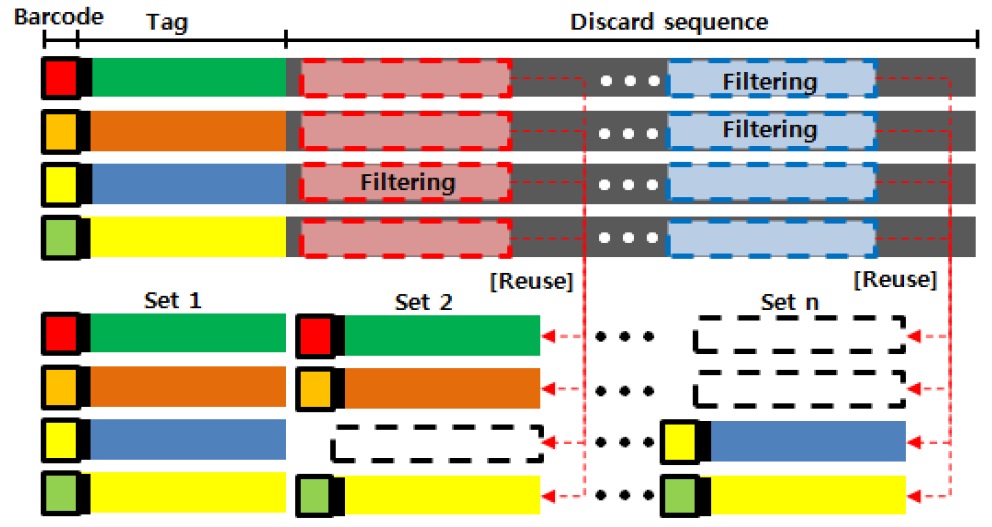
바코드와 제한 효소 정보를 기준으로 통상 오류가 적은 서열의 앞 부분만을 Tag로 추출하여 데이터셋을 생성하고 이를 이용하여 유전형 분석을 하는 TASSEL 방법은 기존 유사 연구들에 비해 비용과 시간을 단축하는 장점이 있다. 반면, 서열의 앞부분만이 Tag로 분석에 사용되기 때문에 그 이후의 데이터는 사용되지 않게된다. 그림 1은 연구에서 사용 될 유전체 서열에 대해서 TASSEL을 이용한 GBS 방법을 수행할 데이터의 구조를 나타낸다.
먼저 FASTQ 파일 포맷으로 이루어진 유전체 서열은 각각 실험적인 정보와 서열에 요약 정보를 나타내는 Header, 유전체 염기의 서열 정보를 가지고 있는 Sequence, 각 염기에 대한 에러율에 대한 정보를 가지고 있는 Quality로 구분되어 있다. 이러한 FASTQ 파일에서 TASSEL을 이용한 분석을 수행할 서열의 구조는 붉은색 박스 부분의 바코드 영역과, 녹색 박스 부분의 앞쪽에 위치한 제한 효소와 태그로 사용될 서열정보가 있다. 서열의 앞에서부터 80번째 이후의 갈색 박스로 표시된 서열이 분석의 사용되지 않는 부분이며 현재 많이 사용되고 있는 NGS 장비들에서 해독되는 서열의 길이가 150 bp 임을 고려할 때 절반 가량을 차지하게 된다.
본 논문에서는 제안하는 알고리즘을 이용하여 SNP 탐지의 효율성을 높이기 위한 연구 방법을 다음과 같이 설계하였다.
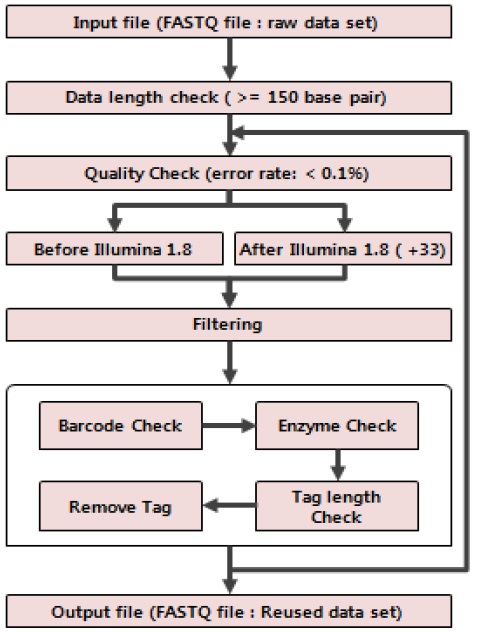
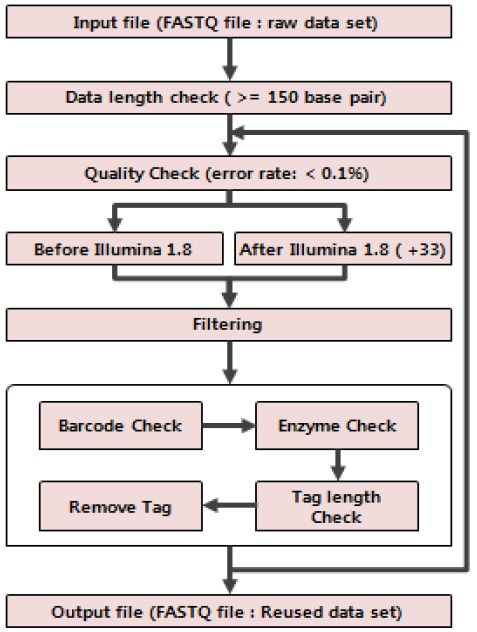
본 연구에서는 GBS 방법에서 분석에 사용되지 않는 유전체 단편 서열들을 재사용하여 SNP 검출 효율을 높이는 연구를 진행하였다. 이에 우리는 그림 2와 같은 순서대로 연구 진행을 위한 설계를 하였다. 먼저 FASTQ 파일 포맷으로 되어 있는 원본 데이터를 입력 데이터로 활용한다. 이러한 원본 데이터는 연구에서 사용되어질 전처리 방법에 적용되기 위해서는 GBS 프로그램인 TASSEL의 바코드와 제한 효소 그리고 tag의 길이를 고려한 특성상 최소 150base pair(bp) 이상이 되어야 한다. 따라서 우리는 150bp이상 길이를 가진 원본 데이터를 체크 하였다. 또한 결과 데이터의 신뢰성을 높이기 위해 염기서열의 에러율 0.1% 이하 데이터를 체크하여 분석에 사용하였다. 이렇게 퀄리티 체크를 수행한 후 유전체 서열을 생산한 장비에 따라 퀄리티 스코어 계산방법을 고려하여 에러율을 충족시키지 못하는 서열들을 제외하는 방법으로 필터링을 하였다. 이렇게 얻은 퀄리티 좋은 데이터를 바코드 인식과, 제한 효소 검사 그리고 태그 길이를 확인한 후 GBS에서 사용 될 서열의 길이만 잘라 최종적으로 FASTQ 파일 포맷으로 되어 있는 데이터 셋을 생성한다. 이러한 작업 절차를 유전체 서열의 길이를 고려하고 반복 수행함으로서 GBS에서 사용될 데이터 셋을 충분히 확보 할 수 있었다.
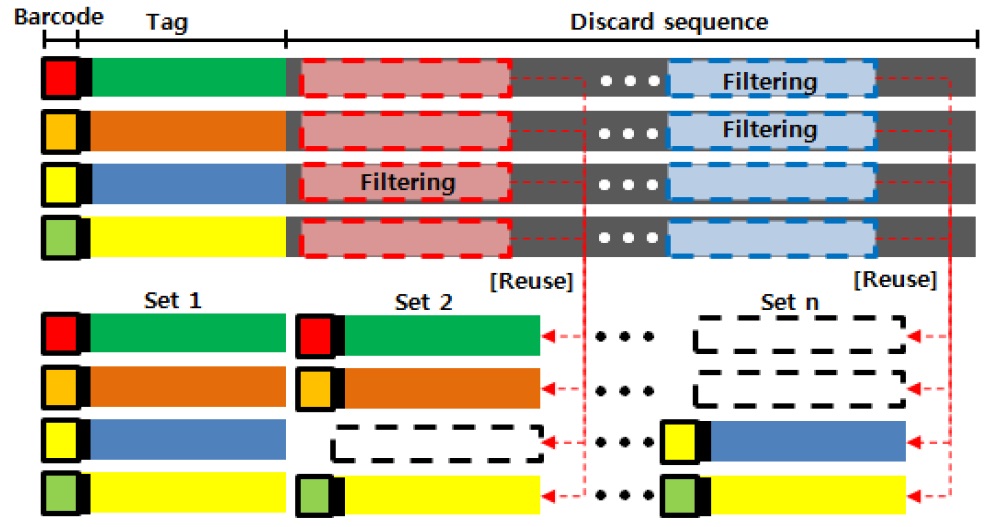
본 연구에서 제안하는 GBS 방법 중 사용되지 않는 유전체 서열을 재사용하는 방법에 대한 그림이다. 기존 TASSEL 방법에서는 유전체 서열정보에서 바코드와 제한 효소를 인식하고 사용할 일정 길이를 추가적으로 잘라내 데이터 셋으로 활용하였다. 하지만 본 연구에서는 사용되지 않는 최초 Tag 이후 서열들에 대해서 퀄리티 체크와 필터링을 수행하고 원본 데이터의 바코드와 제한 효소를 계승함으로서 TASSEL 방법에서 사용 할 데이터 셋을 추가적으로 확보하는 방법을 그림 3과 같이 제시한다.
사용되지 않는 유전체 서열 정보를 재사용하여 TASSEL을 이용한 GBS방법에서 추가적으로 데이터 셋을 확보하는 C언어를 사용한 프로그램 소스 알고리즘은 다음과 같다.
Algorithm 1은 데이터 퀄리티 체크에 대한 부분으로 서 사용되지 않는 유전체 서열을 재사용하기 위한 첫단계이다. FASTQ 포맷 구조의 퀄리티 부분에 대한 ASCII 문자들을 for문을 이용하여 하나씩 체크를 하여 에러율에 대한 매트릭스를 이용하여 최종 퀄리티 값을 결정한다.
Algorithm 2는 Algorithm 1에서 결정된 최종 퀄리티 값을 이용하여 해당 시퀀스를 필터링한다. 이렇게 생성된 시퀀스는 퀄리티 값을 고려하여 에러율 0.1% 이하인 내용들을 제외하고 필터링하여 GBS 처리를 위한 데이터로 사용한다.
Algorithm 3은 Algorithm 1과 Algorithm 2에서 필터링하여 에러율이 낮은 퀄리티를 갖는 데이터를 사용하여 데이터 셋으로 만드는 방법이다. 각각 퀄리티 체크한 데이터에서 바코드 인식과 제한 효소 체크를 한 후에 일정 길이를 잘라낸다. 자른 데이터를 제외한 나머지 시퀀스 데이터에 이전 데이터 셋에서 사용된 바코드와 제한 효소를 각 시퀀스 조각에 승계하여 새로운 데이터 셋으로 생성한다.
이렇게 생성된 새로운 데이터 셋은 기존 TASSEL에서 사용되지 않는 데이터로 만들어진 데이터로 GBS의 최종 결과물인 단일 염기 다형성 탐지하는데 효율을 증가 시킬 수 있을 것이라 판단된다.
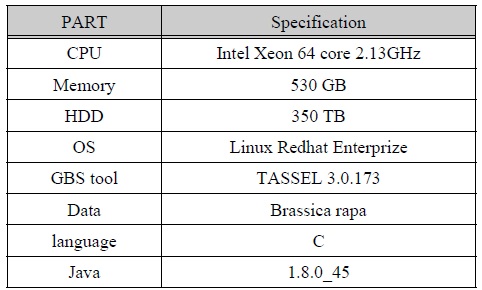
본 논문에서 연구한 시스템의 사양은 다음 표 1 과 같다. Linux Redhat Enterprize 운영체제에서 GBS를 위한 TASSEL 3.0.173을 설치하였다. 또한 Java 1.8.0_45를 설치하여 TASSEL을 구동하였다.
시스템 사양
본 논문에서는 NCBI에서 학명 Brassica rapa(SRA 132035)[10]인 배추과 작물의 데이터를 다운받아 제안하는 알고리즘 성능을 테스트 하였다. 약 251 MB의 참조 유전체 데이터와 150.4 GB 용량의 fastq 포맷 시퀀싱데이터 그리고 실험적인 처리를 통한 바코드에 대한 정보 데이터를 가지고서 TASSEL을 이용한 분석을 진행하였다.
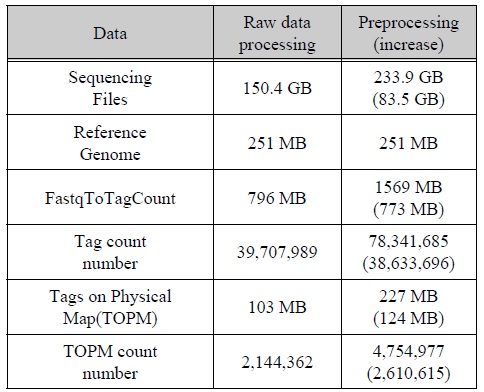
알고리즘을 적용한 전처리 프로그램 적용한 내용과 그렇지 않은 내용을 비교하기 위해 2번에 나누어 표 2와 같이 실험을 진행하였다. 먼저 150.4 GB의 시퀀싱데이터를 통해 TASSEL로 처리하였더니 약 795 MB의 바코드와 인식된 태그들이 생성이 되었다.
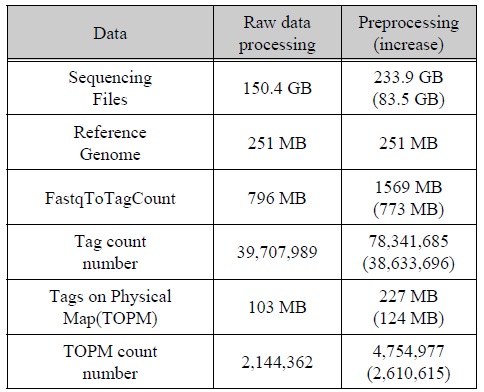
데이터 처리 비교표
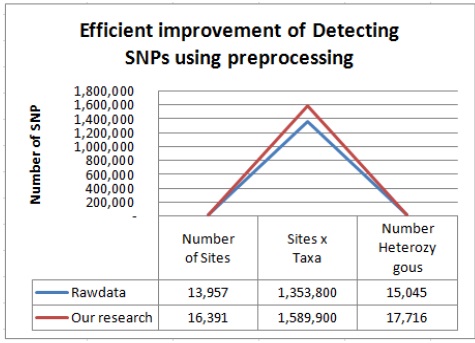
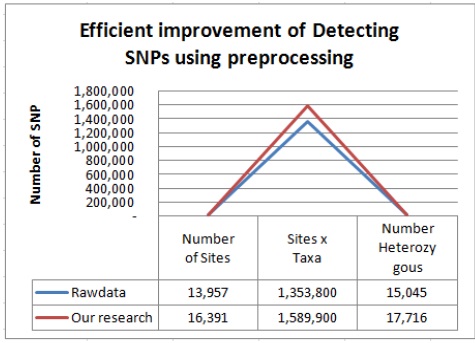
이렇게 생성된 데이터들을 참조 유전체에 맵핑하여 약 103 MB의 데이터를 얻을 수 있었다. 마지막으로 생성된 맵핑 데이터를 토대로 SNP calling작업을 수행하여 총 1,353,800개의 SNP와 15,045개의 헤테로한 SNP을 탐지하였다.
다음으로 우리는 앞서 나온 결과와 비교하기 위해 본 논문에서 제안하는 방법의 알고리즘을 적용하여 실험을 진행하였다. 먼저 150.4 GB의 시퀀싱 데이터에서 뒷부분의 사용되지 않는다고 판단되는 데이터를 재사용하기 위해 퀄리티 체크와 데이터 제거를 통한 새로운 데이터 셋 생성 작업을 진행하였다. 이와같은 작업은 시퀀스의 길이가 150bp로 1회에 퀄리티 체크와 데이터 제거를 통한 새로운 데이터 셋 생성 작업을 수행하였다. 결과적으로 새로운 1개의 데이터 셋을 생성할 수 있었다. 이렇게 생성된 새로운 데이터 셋의 용량은 약 83.5 GB로서 총 233.9 GB의 2개의 데이터 셋을 가지고 GBS 작업을 수행하였다.
먼저 TASSEL로 처리하였더니 기존보다 773 MB 증가한 약 1569 MB의 태그 데이터가 생성되었다. 또한 생성된 태그의 개수가 39,707,989개에서 78,341,685개로 38,633,696개가 증가한 것을 볼 수 있었다. 시퀀스데이터의 용량대비 태그의 늘어난 비율이 틀린 이유는 TASSEL에서 사용되지 않는 데이터의 존재 때문이다. 그에 비해 Preprocessing한 data set에서는 1차적으로 태그의 길이만큼 잘라냈기 때문에 사용되지 않는 데이터가 적다.
이러한 이유에서 용량의 비율과 태그의 개수 비율에 차이가 있다. 태그 데이터를 가지고 참조 유전체 데이터와 맵핑하였더니 124 MB가 증가한 227 MB의 맵핑 데이터를 생성 할 수 있었다.
이러한 데이터를 가지고 SNP calling 작업을 수행하였더니 총 1,589,900개의 SNP와 17,716개의 헤태로한 SNP를 탐지 해 낼 수 있었다. 이와 같은 결과는 기존에 앞부분 만이 사용되고 뒷부분을 사용되지 않는 시퀀스데이터를 재사용하여 그림 4와 같이 전체 약 17.4%정도, 헤테로한 SNP는 17.8%의 SNP를 더 검출 하여 SNP를 탐지하는데 보다 더 좋은 효율을 나타냄을 보여주고 있다.
본 논문에서는 TASSEL을 이용한 GBS 기반 SNP 탐지의 효율적인 개선 방법을 제안 한다. 구체적으로 본 논문에서 제안하는 개선 방법은 기존 프로그램에서 활용되지 않고 사용되지 않는 데이터를 퀄리티 체크와 필터링을 통한 재사용하는 방법으로 사용되지 않는 해당 데이터가 퀄리티 체크와 필터링을 통해 유용한 데이터라 판단되면 분석을 위한 데이터 셋으로 만든다. 이를 통해 기존의 방법보다 더 많은 데이터를 확보 할 수 있다. 또한 이러한 데이터 셋을 통해 TASSEL 방법으로 더 많은 SNP들을 탐지하여 동일한 서열 정보를 이용하여 기존의 방법보다 전체적으로 약 17.4%, 헤테로한 SNP 17.8% 이상의 높은 SNP를 탐지 효율을 보여줌으로서 SNP 관련 연구자들 보다 많은 결과를 얻을 수 있게 해준다.
본 프로그램은 현재 독립형 프로그램으로 소스코드를 다운로드 한 후 컴파일 하여 사용하여야 한다. 이는 컴퓨터에 익숙하지 않은 대다수의 생물학 연구자들에게 어려운 단계로 이를 해결하기 위해 향후에 웹 기반의 서비스[6]를 제공 하고자 한다. 이러한 연구는 유전체를 연구하고 분석하는 사람들에게 보다 효율성이 높은 정보를 제공할 것이다.