Kinsenoside is a principle bioactive compound of Anoectochilus formosanus. It exhibits various pharmacological effects such as antihyperglycemic, antioxidant, anti-inflammatory, immunostimulating, and hepatoprotective activities and has recently been developed as an antidiabetic drug candidate. In this study, as part of an in vitro pharmacokinetic study, the stability of kinsenoside in rat and human liver microsomes was evaluated. Kinsenoside was found to have good metabolic stability in both rat and human liver microsomes. These results will provide useful information for further in vivo pharmacokinetic and metabolism studies.
Kinsenoside [3-(R)-3-β-D-glucopyranosyloxybutanolide] is a principle bioactive compound of
Many compounds with promising pharmacological activities fail to become clinically used drugs because they are rapidly metabolized in the liver or other organs and, consequently, have low bioavailability.
In this study, the metabolic stability of kinsenoside was evaluated using liver microsomal preparations. Kinsenoside concentrations were determined using liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses.
Kinsenoside was generously gifted by Prof. Zhang, Tongji Medical College of Huazhong University of Science and Technology, China. The purity of kinsenoside was >98%, as determined by a high performance liquid chromatography (HPLC) system with an evaporative light scattering detector. HPLC grade acetonitrile was purchased from J.T. Baker (Phillipsburg, NJ, USA). HPLC-grade water was prepared using a Milli-Q purification system (Millipore, Bedford, MA, USA). All chemicals and solvents were of analytical or HPLC grade.
Kinsenoside powder was dissolved in distilled water (DW) at a concentration of 2 mM and further diluted with DW to prepare working standard solutions at concentrations of 10 µM and 100 µM.
>
Stability test in water and buffer
Kinsenoside was incubated in DW or 0.1 M potassium phosphate buffer (pH 7.4) at room temperature at concentrations of 1 µM and 10 µM. At the time points of 0, 1, 2, 4, and 8 h, a 100-µL aliquot was taken and analyzed by LC-MS/MS.
>
Stability test in liver microsomes
The incubation mixture consisted of rat or human liver microsomes (0.5 mg/mL), NADPH generating system (0.1 M glucose-6-phosphate, 10 mg/mL NADP, Glucose-6-phosphate dehydrogenase) and kinsenoside (1 µM and 10 µM) in 0.1 M potassium phosphate buffer (pH 7.4). This mixture was incubated at 37℃ for 0, 10, 30, 60, 90, or 120 min. After incubation, the sample was treated with 200 µL of acetonitrile, vortex-mixed, and centrifuged at 13200 rpm for 5 min. The supernatant was collected and analyzed.
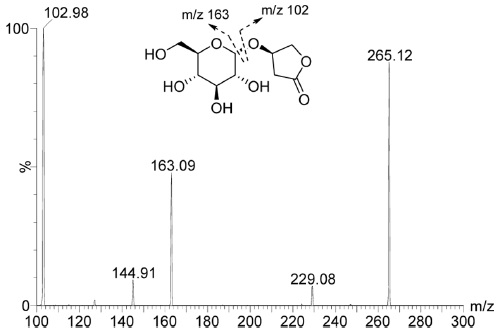
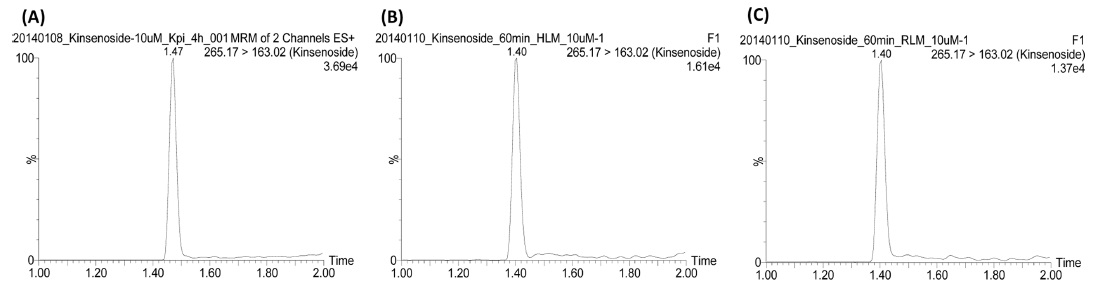
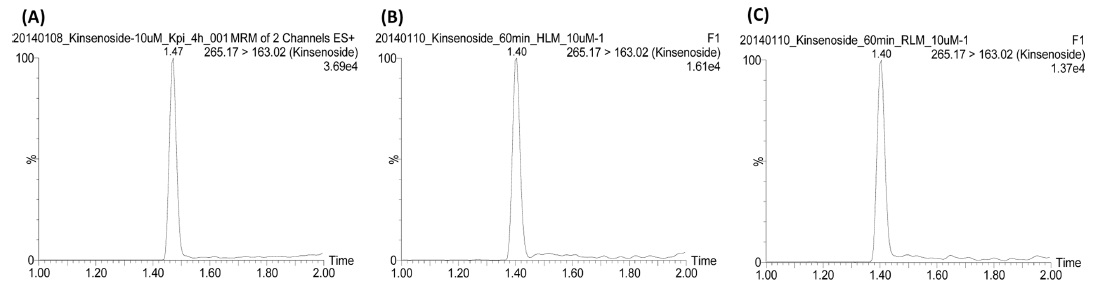
The LC-MS/MS system consisted of Waters Acquity UPLC and Waters Acquity TQD mass spectrometer with an electrospray ionization source. (Waters Corporation, Milford Massachusetts, USA) equipped. The column used for the separation was a Waters Acquity UPLC BEH C18(2.1 mm I.D. × 100 mm, 1.7 µm). The column temperature was maintained constant at 45℃ using a thermostatically controlled column oven. The HPLC mobile phases consisted of 0.1% formic acid in distilled water (solvent A) and 90% acetonitrile in solvent A (solvent B). A gradient program was used for the UPLC separation with a flow rate of 0.2 mL/min. The solvent composition was initially set at 0% B solvent, with gradient elution as; 0-1.5 min, 3%; 1.5-2.0 min, 90%; 2.0-2.5 min maintained 90%; 2.5-2.6 min, 0%; 2.6-4.5 min, 0% of solvent B. Total run time was 4.5 min and injection volume was 3 µL. Mass detection was performed in positive ion mode. The desolvation temperature was 350℃, source temperature was 120℃, and the capillary voltage was 3.5 kV. Nitrogen was used as a desolvation gas at a flow rate of 700 L/h and cone gas at 10 L/h. For multiple reaction monitoring (MRM) analysis, the precursor-product ion pair used was 265.1→163.0 based on the product ion spectrum of kinsenoside (Figure 1). The typical MRM chromatograms of kinsenoside in different reaction solutions were shown in Figure 2.
>
Chemical stability of kinsenoside
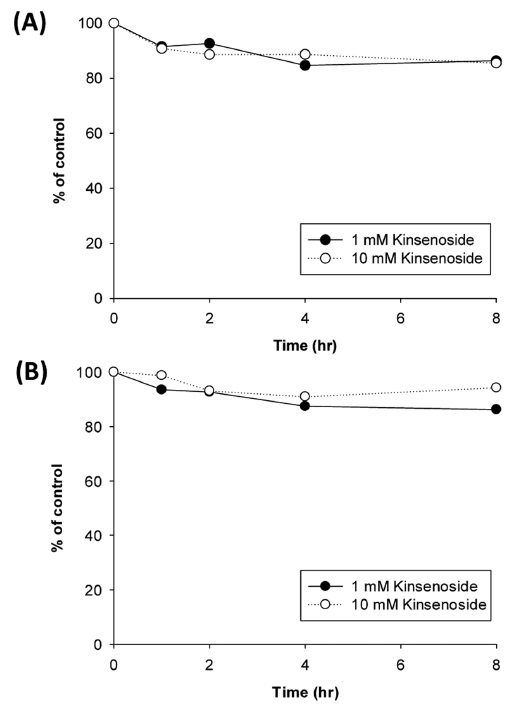
To evaluate the non-enzymatic degradability of kinsenoside, kinsenoside was incubated in distilled water and 0.1 M potassium phosphate buffer (pH 7.4) and the amount of kinsenoside remaining was measured (Figure 3). Kinsenoside was stable in water or buffer solution; the remaining amount of kinsenoside was determined to be 86.3% and 86.2% (versus control value), respectively, after an 8 hincubation of 1 µM kinsenoside at 37℃. Ten micromolar kinsenoside also exhibited the same degree of degradation. These results show that non-enzymatic degradation of kinsenoside does not occur under these conditions.
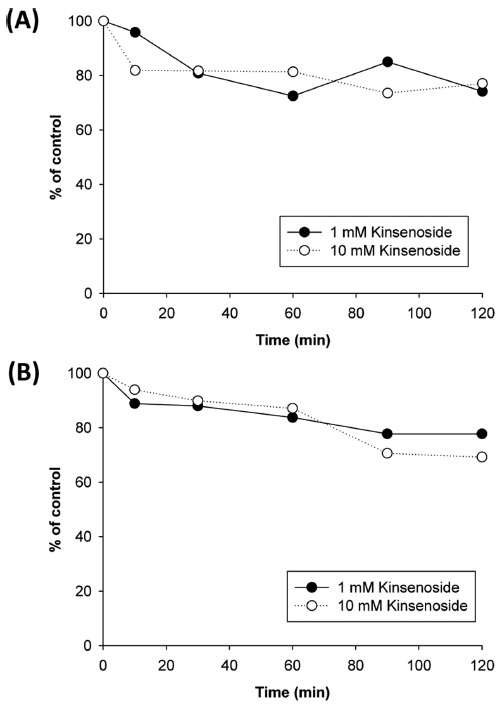
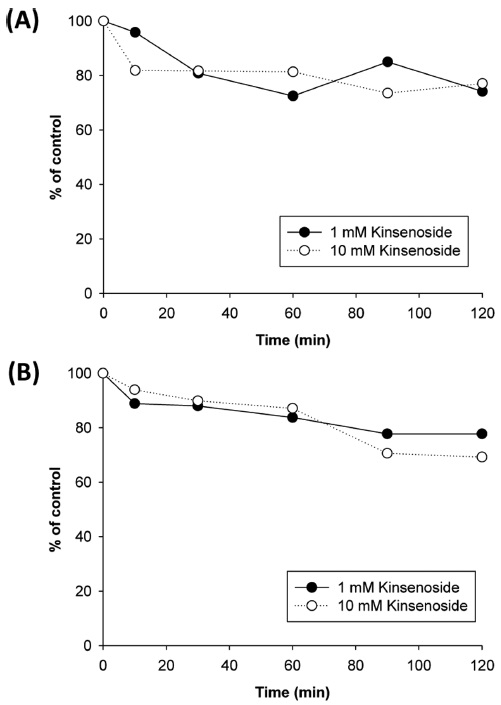
To evaluate the clearance of kinsenoside via phase I metabolism, kinsenoside was incubated with rat and human liver microsomes and the amount of compound remaining was determined at designated time points over 2 h (Figure 4). When kinsenoside was incubated for 2 h at a concentration of 1 µM, the amount remaining was 74.2% and 77.8% in rat and human liver microsomes, respectively. At a concentration of 10 µM, a similar extent of metabolic loss was observed for both species. These data show that kinsenoside is metabolically stable in liver microsomal fractions and the metabolic clearance by hepatic or CYP450 metabolism exerts a minor effect on the total clearance of kinsenoside from the body.
A new antidiabetic drug candidate, kinsenoside, was found to be stable against liver microsomal enzymeassociated metabolism. This result suggests that kinsenoside would be fairly stable and exert pharmacological effects as a parent form rather than active metabolites when used