Recombinant proteins benefit large sectors of the biopharmaceutical, enzyme, and agricultural industries. They are commercially produced in these industries with the aid of genetic and protein engineering. Various systems that produce recombinant proteins, such as bacterial (Lunn
China, Japan, Korea, and India in Asia, and France, Italy and Bulgaria in Europe retain silkworm bioresources in national resource stock centers. By screening silkworm strains maintained at Kyushu University in Japan, Lee
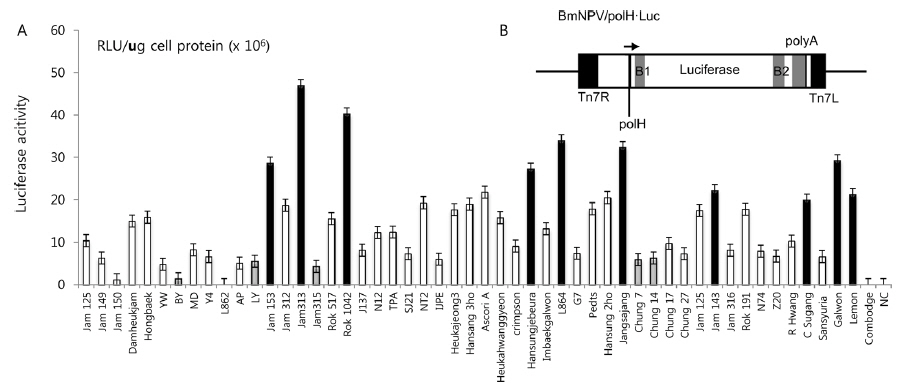
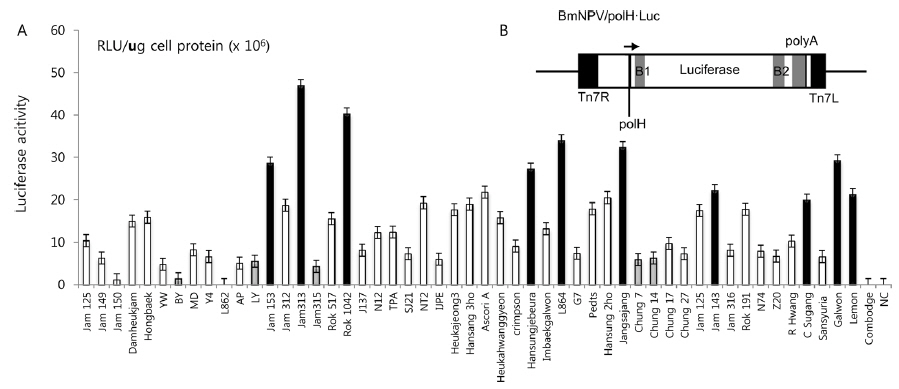
In this study, we screened several silkworm strains by introducing luciferase-expressing recombinant BmNPV, previously constructed by Kawakami
>
Preparation of recombinant baculovirus
In this study, a previously constructed (Kawakami
The experimental silkworm larvae comprised the larvae of 52 strains and, as a control strain, the F1 hybrid strain 125 × 126 (a certified strain used by farmers during summer and fall), supplied by the Gyeongsangbuk-do and the Kangwon-do Foundation Original Seed Production Center (Sericulture and Entomology Experiment Station), respectively. Details regarding these 52 silkworm strains are described in the National Academy of Agricultural Sciences ‘Genebank Information Center' (http:// www.genebank.go.kr/). The nine hybrids used in this study were produced by randomly crossing lines with high-permissive strains that were selected through the third screening in this study and were kept in the Kangwon-do Foundation Original Seed Production Center. Silkworms were reared on fresh mulberry leaves at 25°C–27°C and fifth-instar larvae were injected using a syringe (Hamilton Co., Reno, NV, USA) into the hemocoel, with 20 μL
>
Analysis of luciferase activity
The collected hemolymph was centrifuged at 3,000 rpm for 10 min to remove the supernatant, and the hemocytes were washed in phosphate buffered saline (PBS, pH 7.3) and then resuspended for approximately 10 min in 120 μL lysis buffer containing 25 mM Tris phosphate (pH 7.8), 2 mM 1,2-diaminecyclohexane-N,N,N',N'-tetraacetic acid (DTT), 2 mM 1,2- cyclohexanediaminetetraacetic acid monohydrate (CDTA), 10% glycerol, and 1 % Triton X-100. In total, 100 μL of the lysed solution was added to an equal volume of a substrate solution containing beetle luciferin, and luciferase activity was measured with a GloMax Luminometer (Promega, USA). Data were expressed in relative light units (RLU) per microgram of total cell proteins.
Total RNA was isolated from the fat body of each silkworm strain after the luciferase assay, using TRIzol (Invitrogen, Grand Island, NY, USA), and eluted with RNase–free water. For semi- and quantitative reverse-transcription PCR (RT-PCR) analysis, 0.5 μg total RNA from each silkworm strain was reverse transcribed into cDNA using a first cDNA synthesis kit (Toyobo, Japan). The 20 μL reaction mixture, containing 0.5 μg total RNA, 4 μL 5X reverse transcriptase buffer, 2 μL 10 mM dNTPs, 1μL 10 U/μl RNase inhibitor, 1 μL 10 pmol/μL oligo (dT)20 primer, and 1μL ReverTra Ace® reverse transcriptase, was incubated at 42°C for 20 min. Subsequently, 1μL of each firststrand cDNA product was used for semi- and quantitative realtime RT-PCR. Each 50μL reaction mixture of semi-RT PCR contained 0.5 μL 1st cDNA, 0.25 μL 5 U/μL TaKaRa Ex Taq polymerase (Takara Bio Inc., Japan), 5 μL 10X Ex Taq buffer, 4 μL dNTP mixture, and 10 pmol each 5′ and 3′ primers. The thermal cycling profile was as follows: 94°C for 5 min, 27 cycles of 94°C; for 30 s, 57°C for 30 s, and 72°C for 30 s, followed by a final extension at 72°C for 5 min (Takara Bio Inc., Japan). Each PCR product was electrophoresed in a 1.5% agarose gel in Tris acetate EDTA (TAE) buffer. Next, real-time PCR was performed according to SYBR Premix Ex Taq (Takara Bio Inc., Japan) in the Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems, Grand Island, NY, USA). The thermal cycling profile was as follows: denaturation at 94°C for 30 s, followed by 40 cycles of 95°C for 5 s, 60°C for 34 s. The
The recovered fat bodies (0.3 g fat body can be harvested from each silkworm larva) were suspended in 1 mL homogenization buffer (0.5 M Tris buffer containing 0.5 M NaCl, 1% NP40, 5% complete, pH 8.0) and homogenized using a Vibra-Cell homogenizer (Sonics, USA) for 5 min at °C. The homogenate was ultracentrifuged at 14,000 rpm for 20 min at 4 °C, the supernatant was re-centrifuged and used for western blotting and ELISA analysis. The concentrations of the purified proteins were determined using the bicinchoninic acid (BCA) method, using bovine serum albumin as a standard (Smith
The fat body samples from silkworm larvae were denatured in 4x Laemmli sample buffer and resolved on 10% SDS-PAGE. After electrophoresis, proteins were transferred onto PVDF membranes for western blotting (Millipore, USA) and the membranes were blocked for 1 h at room temperature (RT) in TBS-0.1% Tween 20 (TBST) with 5 % skim milk, washed with TBST, and then incubated at room temperature for 1 h using antiluciferase antibody (Rochland, USA). The membranes were then washed five times with TBST, and incubated with anti-goat HRP conjugate (Rochland, USA) for 1 h. After extensive washing with TBST, protein bands were detected using the ECL western blotting detection system on Hyperfilm ECL Films (Amersham, USA).
The luciferase expressed in silkworm larvae was quantified using the ELISA indirect protocol modified (Abcam, USA) with luciferase antibody. First, the diluted proteins from silkworm fat body were added to a Nunc plate (Thermo Scientific, USA), incubated for 1 h at room temperature (RT) and washed three times with TBST. A total of 200 μL TBST with 1 % bovine serum albumin (BSA) as blocking solution was added to the protein-coated plate wells, followed by incubation for 30 min at RT. The wells were washed three times with TBST. Then, 100 μL per well of 10 μg/mL firefly anti-luciferase solution were added as a coating antibody, the wells were incubated for 1 h at RT and then washed with TBST. As a second antibody, 100 μL goat-HRP conjugate diluted 5,000 times in TBST were added to each well, incubated for 1 h and washed three times with TBST buffer. A total of 100 μL of 3,3′,5,5′- tetramethylbenzidine substrate (TMB in 100 mM citric acid with 0.02 % of hydrogen peroxidase) was added to each well and left at RT for blue color development (KPL, USA). The reaction was stopped by the addition of 100 μL of 2 M H2SO4 solution. The optical density (OD) of the samples was measured at a wavelength of 450 nm. Firefly luciferase reference serum (Sigma, USA, 1 mg/mL) was used as a standard for the calibration of luciferase.
>
Selection of a high-permissive silkworm strain as a bioreactor by use of BmNPV
In previous studies, Lee
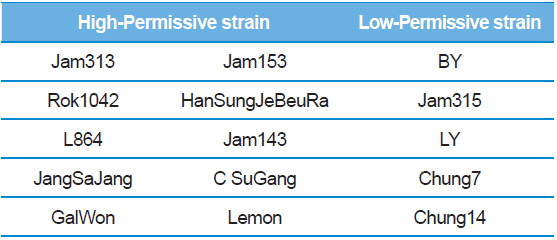
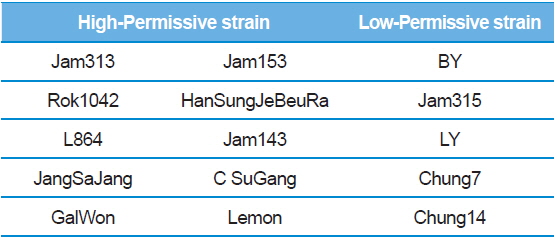
Summary of the silkworm host showing high- and low-activity of BmNPV/polH·Luc in silkworm used in this study
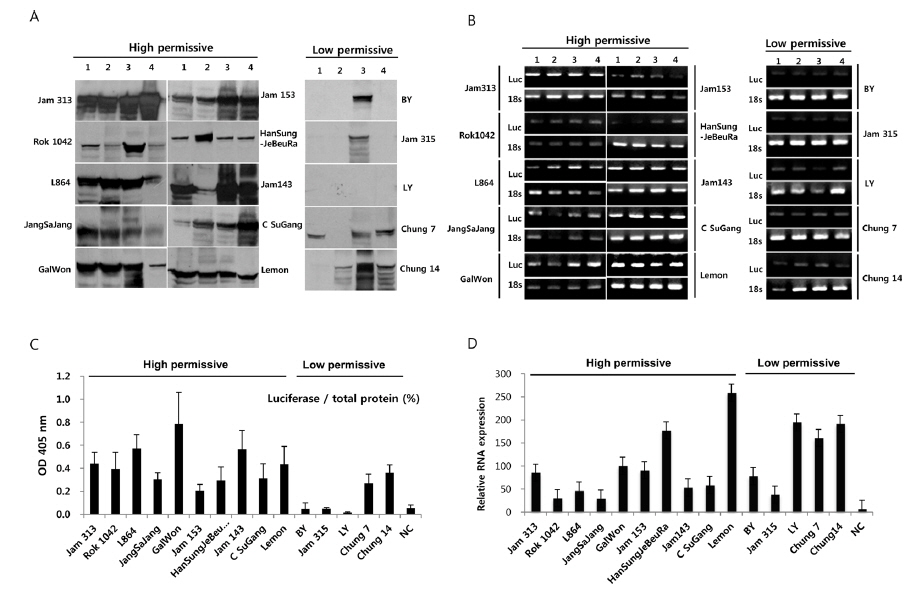
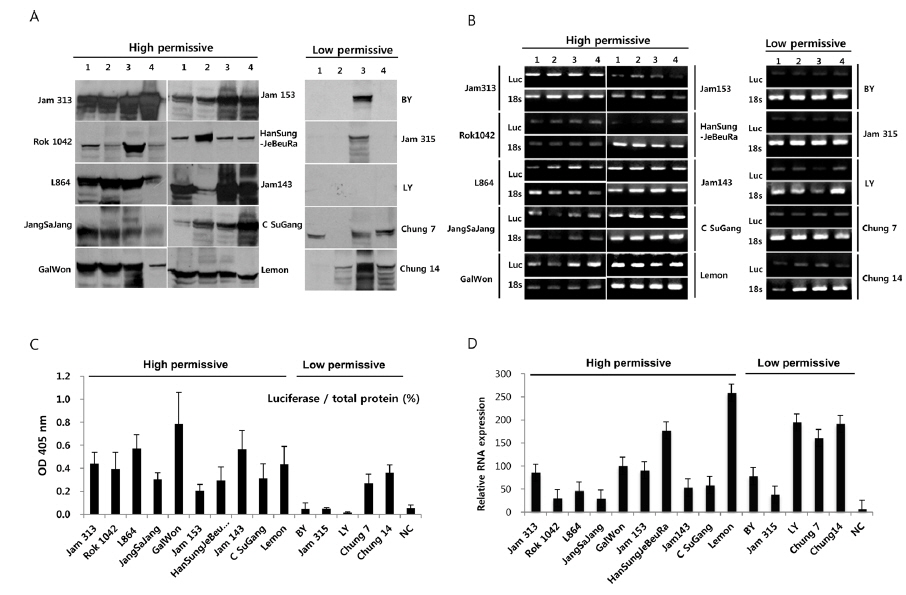
To further confirm the luciferase activity in selected high- and low-permissive strains, crude preparations of individuals (n = 4) of each strain were analyzed via 10% SDS-PAGE and subjected to western blot (Fig. 2A) and ELISA (Fig. 2C) analysis using a firefly luciferase antibody. As shown in Fig. 2A, 10 high-permissive strains (left panels) showed higher expression levels of the luciferase protein, isolated from larval fat body, than 5 low-permissive strains (right panel). The expression of luciferase was confirmed through detection of a predicted band size of around 62 kDa. It was further confirmed by ELISA analysis by using the same luciferase antibody (Fig. 2C). The expression level of luciferase in all 10 high-permissive strains was higher than that in the low-permissive group, and, as expected, results derived from both luciferase activity and western blot analysis were found to be of a highly similar pattern (Fig. 2A, Fig. 2B, and Fig. 2C), while individuals of each strain exhibited slight variations in their level of luciferase expression.
To characterize the level of luciferase gene transcription, we employed semi-quantitative and quantitative real time PCR (Fig. 2B and 2D). The results showed no significant correlation between the level of luciferase gene transcription and the allocation of strains to the high- and low-permissive groups. Luciferase gene transcription levels of HanSungJeBeuRa and Lemon as high-permissive strains and of LY and Chung14 as low-permissive strains were much higher than that of Jam313, which had shown the highest luciferase activity (Fig. 2D). Whether levels of transcription and translation fluctuate in their correlation remains to be discovered. Further reciprocity studies using the recombinant counterparts may clarify the issue.
>
Comparison of luciferase yields in purebred and hybrid strains
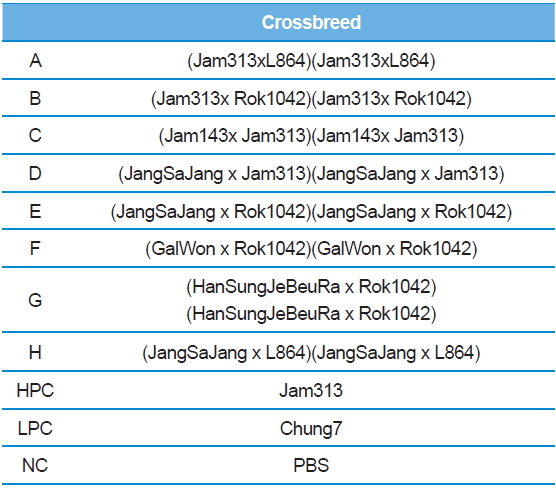
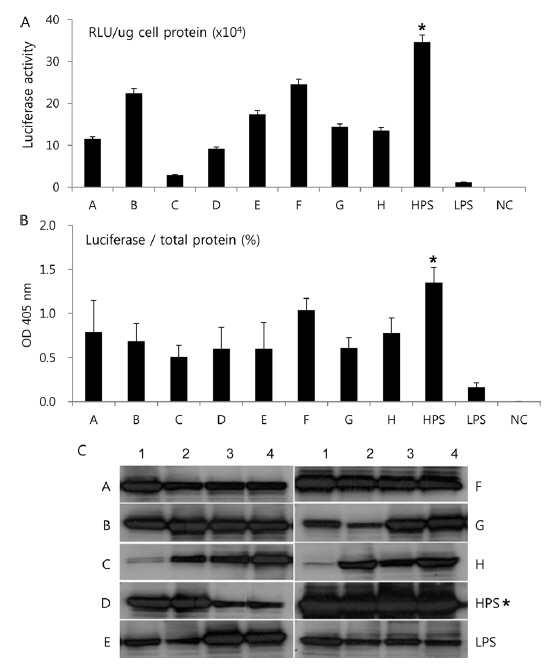
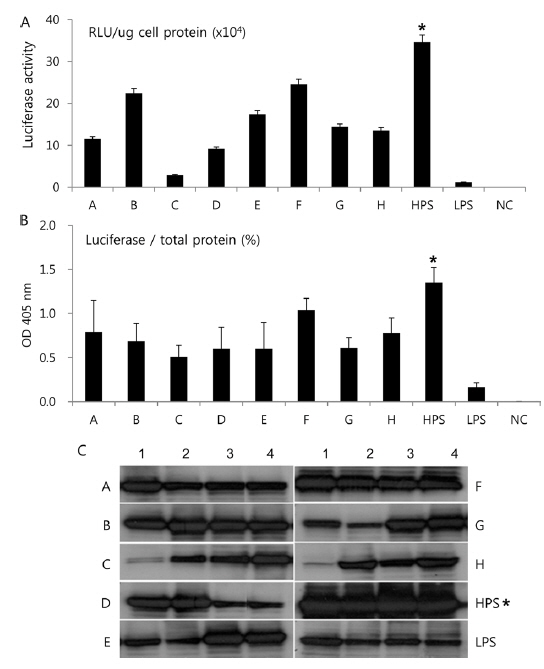
In Korea, the commercially available strains, such as Jam123 x Jam124, Jam125 x Jam126, Jam125 x Jam140, etc., are crossed at the Foundation Original Seed Production Center annually and then widely used by sericultural farm households. Because of the genetic weakness of pure breeds, we conducted F1 hybrid and three-way crossing using highpermissive strains for supply convenience. By three-way crossing, 9 combinations of high-permissive strains were prepared with the high-permissive strain Jam313 and the low-permissive strain Chung7 as control strains (Table 2). Among the 10 high-permissive strains, we hybridized using Jam313, Rok1042, L864, JangSaJang, HanSungJeBeuRa, and GlaWon with high levels in luciferase activity. Three-way combination strains were lower in luciferase activity than Jam313, tested as high-permissive control (HPC), and significantly higher in luciferase activity than Chung7, tested as low-permissive control (LPC, Fig. 3A). The luciferase expression was further investigated in 9 three-way combination strains, high-, and low-permissive controls, by western blot and ELISA (Fig. 3B and 3C). Both methods clearly demonstrated that the luciferase expression level was high in Jam313 and low in Chung7, and these results are in agreement with the respective luciferase activities (Fig. 3). Previous reports have shown that the F1 hybrid or three-way cross of the
[Table 2.] List of three ways cross produced by random combinations with high-permissive strains
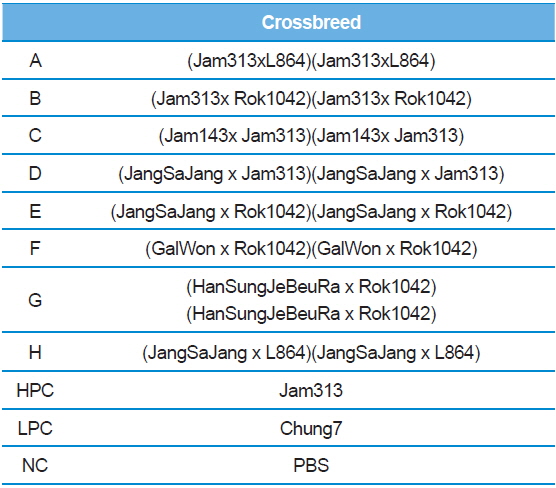
List of three ways cross produced by random combinations with high-permissive strains
This study suggested that meticulous selection of a more suitable strain is required for more efficient production of heterologous protein, through the application of recombinant bacmid DNA derived from domestic