We previously identified Streptomyces griseus as an anti-cancer agent (
The marine environment is a rich source of natural products with a wide variety of biological activity. During the last three decades, more than 15,000 natural products were isolated from marine organisms (MarinLit, 2008). In many cases, pharmaceutically interesting natural products were isolated from microbes associated with marine invertebrates, rather than the marine invertebrates themselves (Faulkner et al., 2000; Haygood et al., 1997; Proksch et al., 2002; Thoms et al., 2005a; Thoms et al., 2005b). Sponges harbor significant amounts of bacteria in their tissues. In some cases bacteria make up more than 40% of the sponge biomass (Vacelet, 1975; Bewley and Faulkner, 1998). Marine sponge-associated microbes rely on sponges for nutrient acquisition and secondary metabolite production (Hentschel et al., 2002). The various secondary metabolites synthesized by microbial associates also possess excellent bioactivity in many studies (Pimentel-Elardo et al., 2010; Liu et al., 2005; Bringmann et al., 2003, 2005; Zheng et al., 2005). Thus, sponge-associated micro-organisms represent an attractive source of marine natural products.
By employing sophisticated techniques and various screening programs, 22,500 biologically active compounds were extracted from microbes. Among these, 45% were from Actinobacteria, 38% from fungi, and 17% from unicellular bacteria. Importantly, Actinobacteria, particularly
To continue our work isolating and evaluating the biological activity of microbial metabolites, we identified active compounds exhibiting an anticancer effect in Actinobacterium strain,
RPMI-1640 medium, fetal bovine serum (FBS), penicillin-streptomycin, and trypsin-EDTA were purchased from Gibco/BRL (Burlington, ON, Canada). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), RNase A, Dihydroethidium (DE), 2’ 7’-dichlorodihydro fluorescein diacetate (DCFH2-DA), propidium iodide (PI), dimethyl sulfoxide (DMSO), and Hoechst 33342 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against Bax, Bcl-xL, cleaved caspase-3, poly-ADP-ribose polymerase (PARP) and GAPDH were purchased from Cell Signaling Technology (Bedford, MA, USA). All solvents used for preparation of crude sample and CPC separation were of analytical grade (Daejung Chemicals &Metals Co., Seoul, Korea). HPLC grade solvents were purchased from Burdick & Jackson (MI, USA).
>
Culture and Extraction procedure of Streptomyces griseus
2 L Erlenmeyer flasks containing 2 L of medium containing of peptone (0.7%), yeast extract (0.2%), D-(+)-Glucose (0.2%) and seawater (100%) were inoculated with a single colony from a well grown agar plate. The bacterial strain was cultured (18 L) for 20 days (static) at 29℃ with humidity. The cultured broth of the strain
>
Separation procedure of centrifugal partition chromatography (CPC)
CPC operation was performed by modified protocol of Lee et al. (2014). LLB-M high performance CPC 240 (Sanki Engineering, Kyoto, Japan) was used in preparative CPC. The EtOAc extract was suspended in H2O and fractionated with
>
HPLC-DAD and HPLC-DAD-ESI/MS analysis
HPLC (Waters, Milford, Massachusetts, USA) and HPLC-DAD-ESI/MS (Hewlett-Packard 1100 series, Waldbronn, Germany) operation were performed by modified protocol of Lee et al. (2014). Gradient was from 5% to 50% for solvent A in 50 min, from 50% to 100% for solvent A in 10 minutes with a 10-min hold at 100% for solvent A. Multiple wavelength monitoring was performed at 210, 254, 280 and 365 nm and photodiode array detector measured from 200 to 400 nm. Operating HPLC-DAD -ESI/MS, negative ion mass spectra of the column eluate were recorded in the range m/z 100-2000.
>
1H-NMR and 13C -NMR analysis of purified compounds
The 1H NMR and 13C NMR spectra of the isolated compounds were recorded on a JEOL JNM-ECP 400 MHz NMR spectrometer, using Methanol-
HL-60 (Human promyelocytic leukemia cell line) was maintained at 37℃ in an incubator with humidified atmosphere of 5% CO2. Cells were cultured at a concentration of 5 × 104 cells/ml in RPMI-1640 medium supplemented with 10% (v/v) heat-inactivated FBS, penicillin (100 U/mL) and streptomycin (100 μg/mL) for further experiments.
>
Cell growth inhibitory assay
The cell growth inhibitory assays of the test samples were examined by MTT assay and modified protocol of Kang et al. (2012). A 5 × 104 of HL-60 cells was seeded in 96-well plate, and then the cells were treated with 10 μL of the samples, and further incubated for 36 h. And then was measured via ELISA at a wavelength of 540 nm by MTT stock solution (2 mg/mL).
>
Nuclear staining with Hoechst 33342
The nuclear morphology of cultured HL-60 cells was studied using modified protocol of Kang et al. (2012). The cells with homogeneously stained nuclei were considered viable, whereas the presence of chromatin condensation and/or fragmentation was indicative of apoptosis (Gschwind and Huber, 1995; Lizard et al., 1995). HL-60 cells were seeded into 24-well plates at a density of 1 × 105 cells/mL. The cells were then treated with various concentrations of the target compound and incubated for an additional 36 h. Then, Hoechst 33342 and PI, a DNA specific fluorescent dye was added to the culture medium at a final concentration of 10 and 5 μg/mL, respectively. The stained cells were then observed under a fluorescence microscope equipped with a CoolSNAP-Pro color digital camera to determine the degree of nuclear condensation.
Cell cycle analysis was measured using modified protocol of Kang et al. (2012). The HL-60 cells were seeded into 5 cm dishes at a density of 1 × 105 cells/mL. The cells were then treated with the target compound and incubated for 36 h. Flow cytometric analysis was conducted with an FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). The effect of the target compound on the cell cycle was determined by changes in the percentage of cell distribution at each cell cycle phase, and assessed by histograms generated by the Quest and Mod-Fit computer programs (Wang et al., 1999).
The accumulation of intracellular H2O2 was measured modifying previously described protocol (Wen et al., 2002; Zhang et al., 2003). In brief, the HL-60 cells were placed in 6-well plates at a concentration of 5 × 104 cells/mL. The cells were treated with various concentrations of the target compound. After 48 h the fluorescence was analyzed using a flow cytometer (Becton Dickinson, San Jose, CA, USA).
Western blot analysis was performed by modified protocol of Kang et al. (2012). Cells (1 × 105 cells/mL) were treated with various concentrations of the target compound and harvested. To measure apoptosis mechanism, primary antibody against Bax, Bcl-xL, cleaved caspase-3, PARP, and GAPDH were used. Signals were developed using an ECL western blotting detection kit (WESTAR C ULTRA, CYAGEN, Italy) and exposed to X-ray films.
>
Origin and maintenance of zebrafish
Adult zebrafish were obtained from a commercial dealer (Seoul Aquarium, Korea) and 10 fish were kept in a 3-L acrylic tank under the following conditions: 28.5 ℃, with a 14/10 h light/dark cycle. Feeding the fish was performed as protocol of Ko et al. (2014).
>
Evaluation on toxicity of PA in zebrafish
For the toxicity of PA in zebrafish embryos heartbeat rates, cell deaths and survival rates in zebrafish embryos were measured according to protocols by Ko et al. (2014). The images of stained embryos were observed using a fluorescence microscope (CKX41, Olympus, Japan), which was equipped with a CoolSNAP-Pro colour digital camera (Olympus, Japan).
The Student’s
>
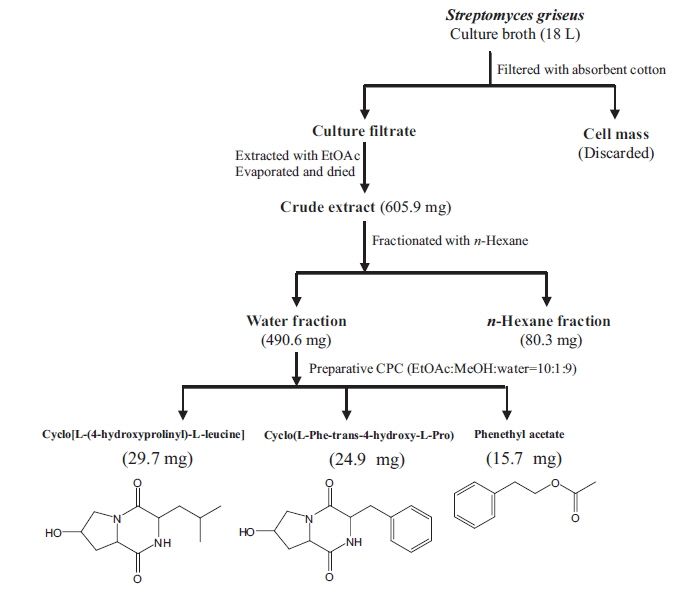
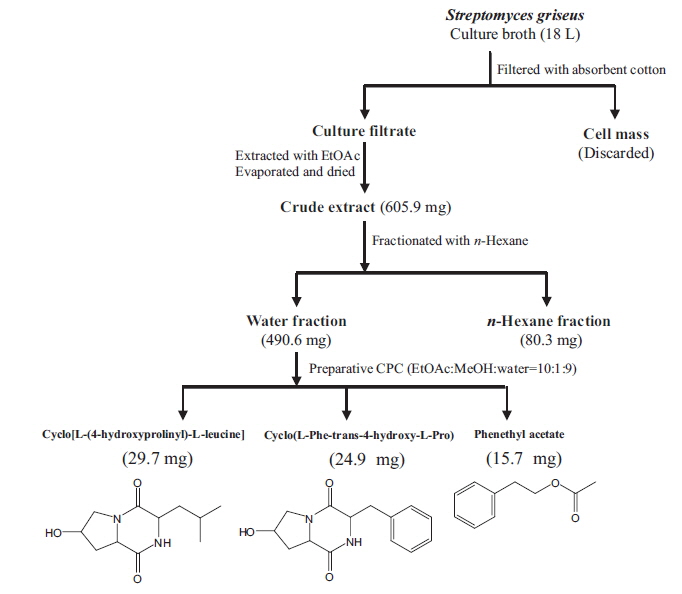
Isolation of main compounds from S. griseus extract
We isolated bioactive metabolites from
Cyclo[L-(4-hydroxyprolinyl)-L-leucine]: pale yellow liquid, 1H NMR (Methanol-
Cyclo(L-Phe-trans-4-hydroxy-L-Pro): pale yellow liquid, 1H NMR (Methanol-
Phenethyl acetate: pale yellow liquid, 1H NMR (Methanol-
>
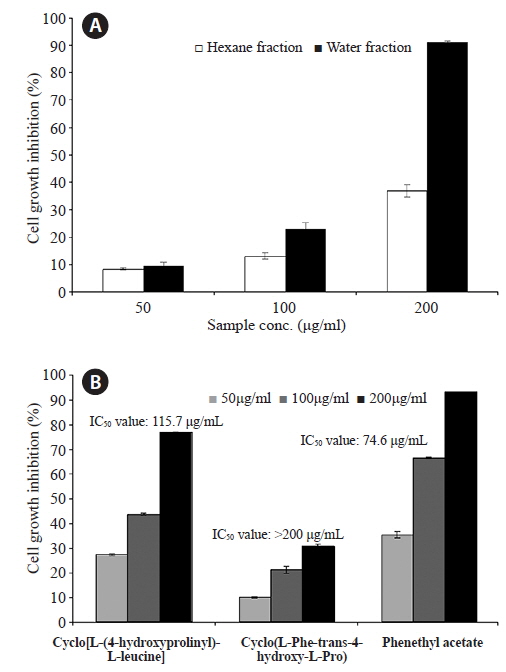
Anti-proliferative effects in HL-60 cells
The dried
>
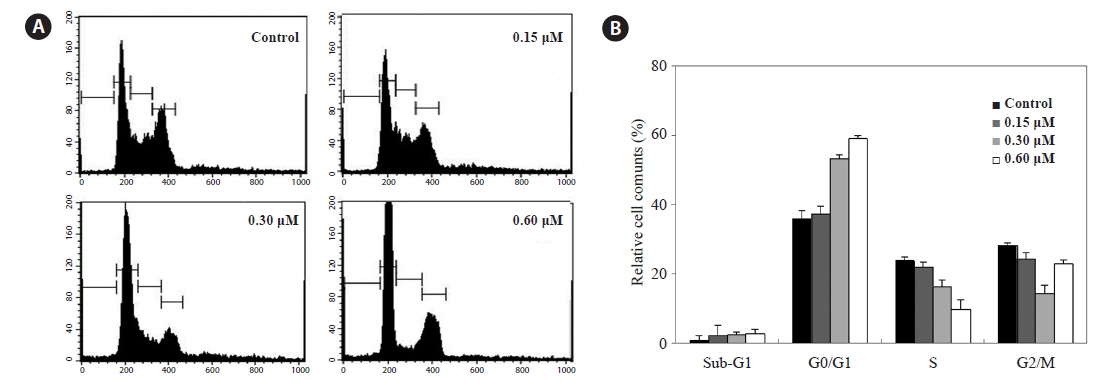
Anticancer activity of PA via cell-cycle arrest
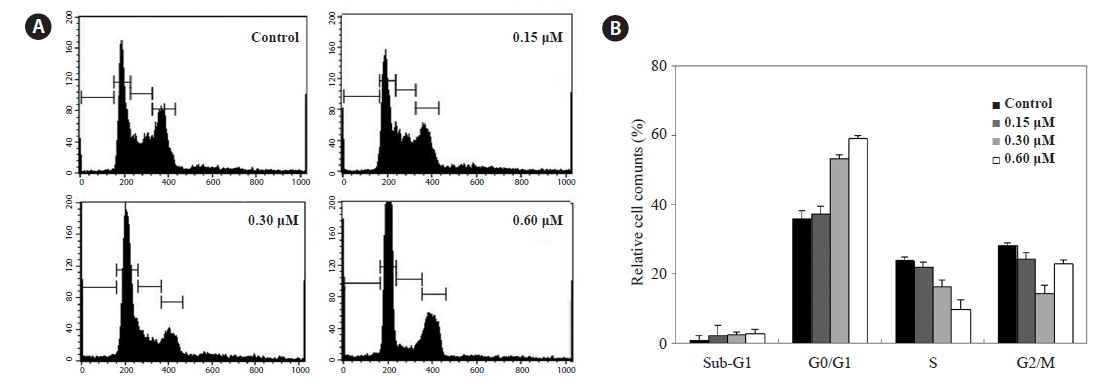
Because HL-60 cells are highly sensitive to PA, we examined whether PA could interfere with the cell cycle by flow cytometry. HL-60 cells were incubated with PA (0.15, 0.30 and 0.60 μM) for 48 h analyzed by flow cytometry. PA decreased the proportion of cells in the S and G2/M phases, and dose-dependently, increased those in G0/G1 phase (Fig. 3A). Furthermore, the proportion of cells in the sub-G1 phase, which is associated with apoptosis, slightly increased.
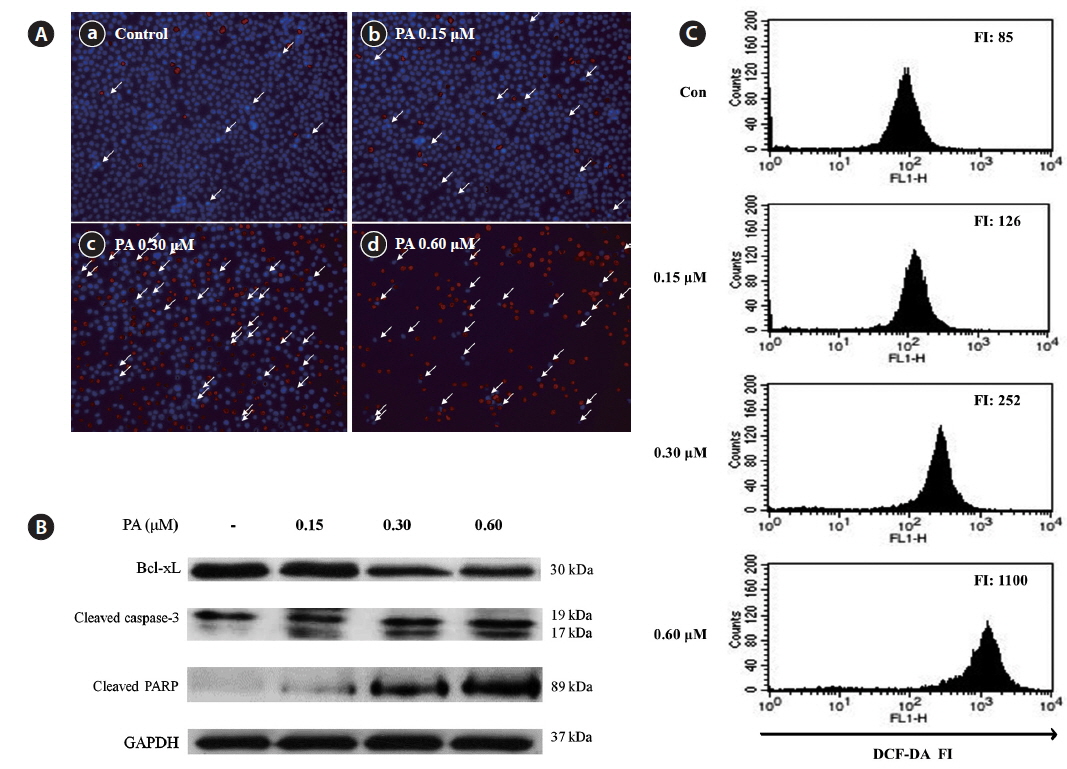
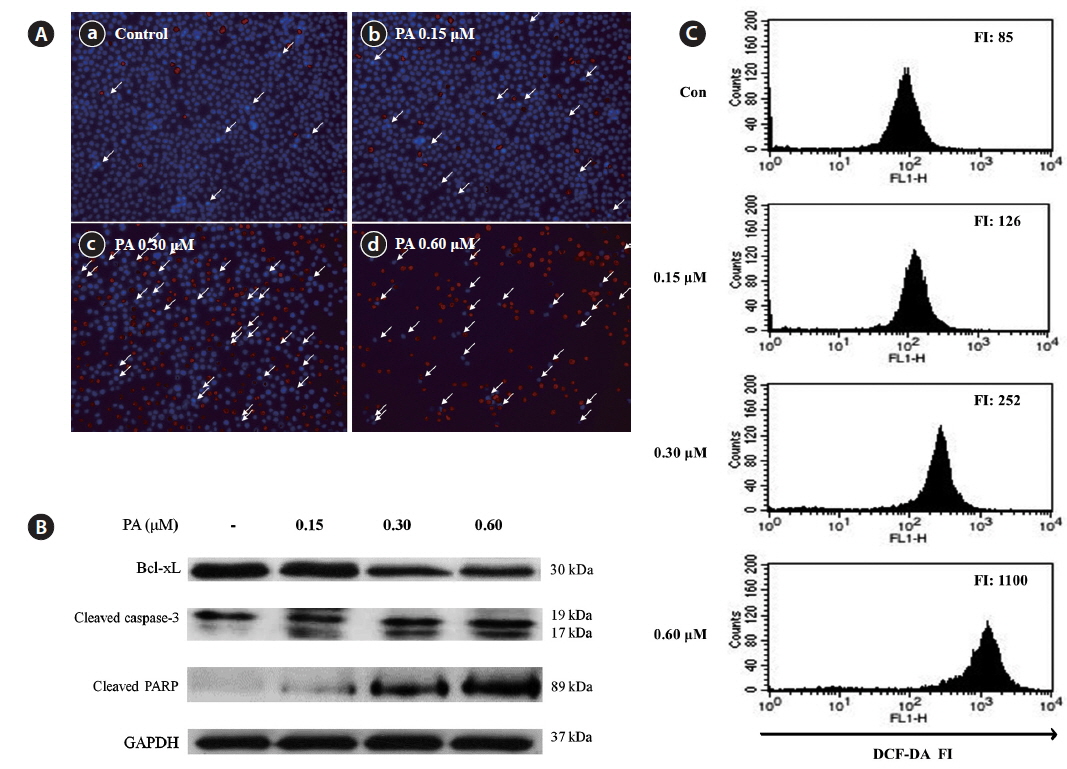
Induction of cell death and apoptosis by PA was further studied using a Hoechst-PI double staining assay (Fig. 4A). Untreated control cells exhibited no DNA damage; however, obvious cell damage was observed in the PA-treated cells. PA dramatically increased both the number of apoptotic bodies and cell death in HL-60 cells. To evaluate the mechanism of PA-induced apoptosis, the expression of pro-apoptotic (Bax, PARP and caspase-3) and anti-apoptosis (Bcl-xL) proteins were evaluated by western blot (Fig. 5B). The expression level of Bcl-xL decreased in a dose-dependent manner. In contrast, cleaved PARP was dramatically increased, whereas cleaved caspase-3 was slightly increased by PA treatment.
We evaluated whether PA induces apoptosis via ROS accumulation in HL-60 cells. DCFH-DA was used to measure total ROS production in cells (Fig. 4C). PA treatment dose-dependently increased ROS production in HL-60 cells. In particular, the fluorescence intensity increased 13-fold when cells were treated with 0.60 μM PA, as compared to the control. These results suggest that PA-induced ROS generation in HL-60 cells may be important for the anticancer effects of PA.
>
PA toxicity in vitro and in vivo
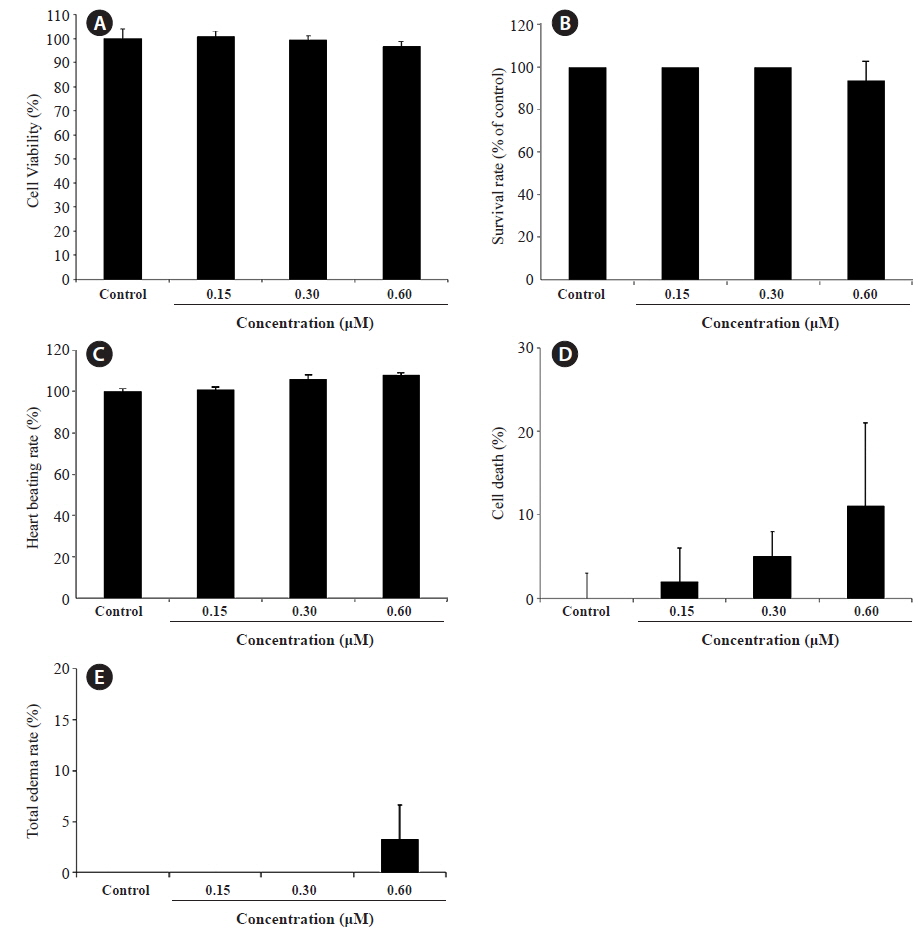
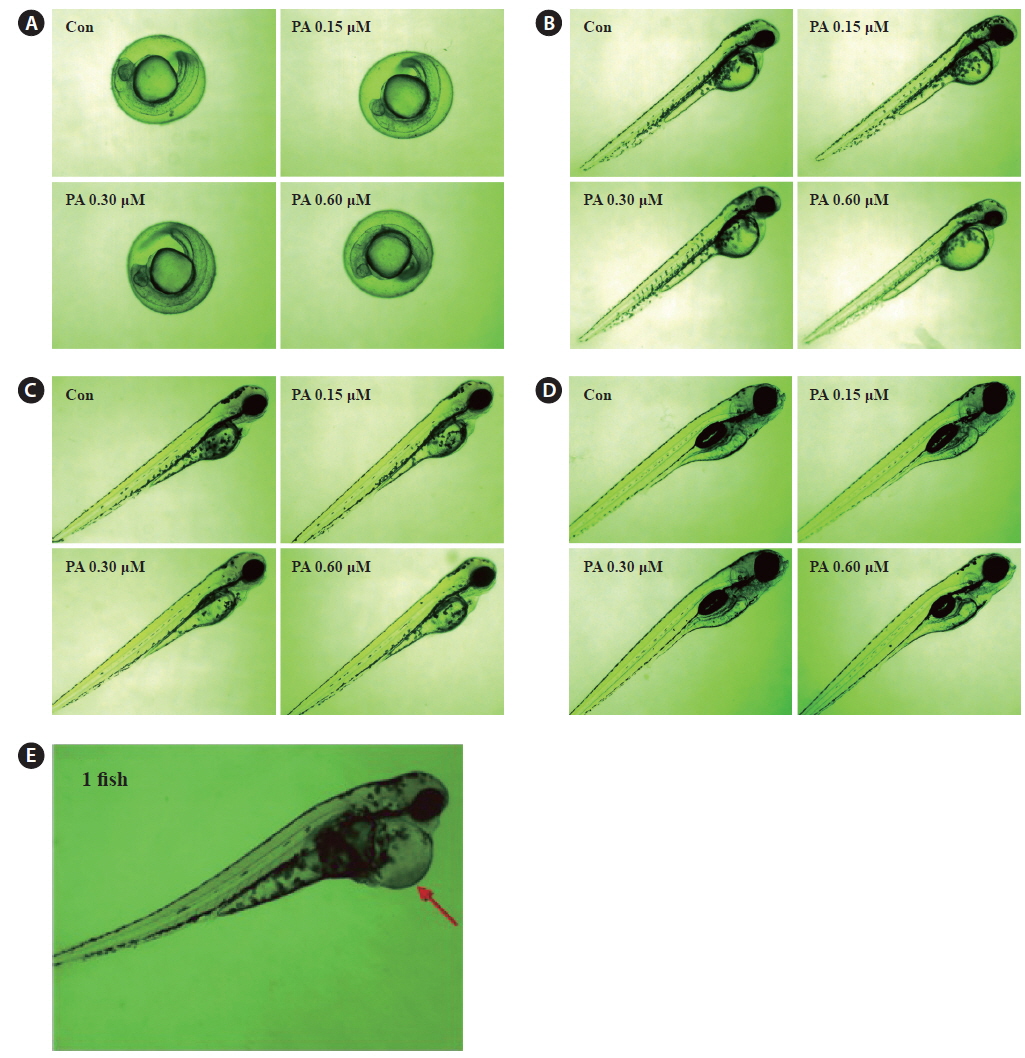
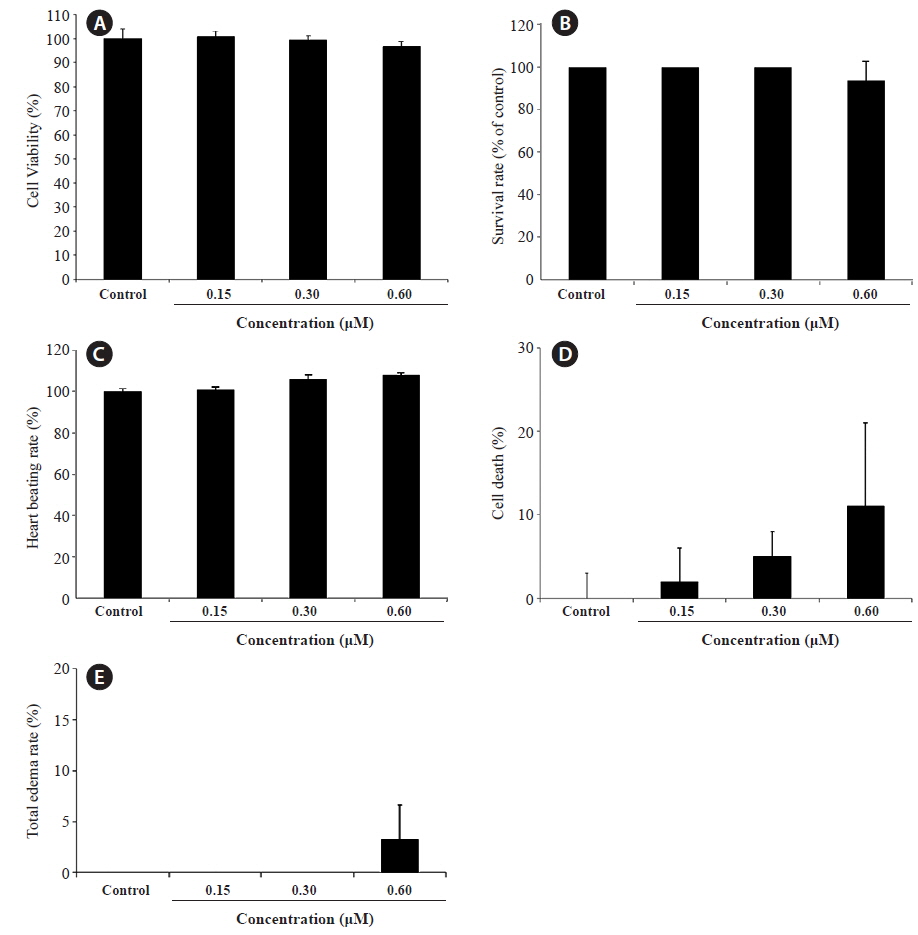
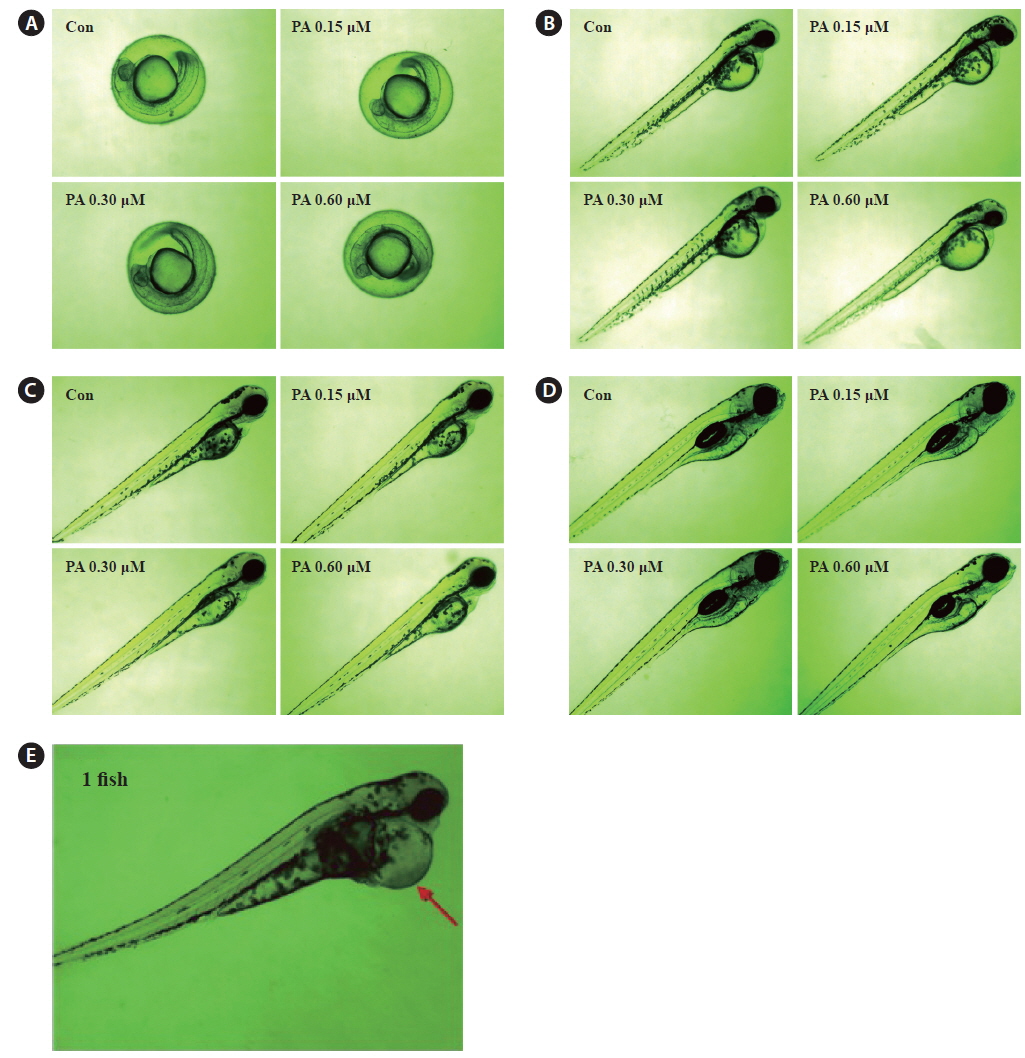
We evaluated the cytotoxicity of PA in Vero monkey kidney cells (Fig. 5A), and monitored the survival (Fig. 5B) and growth patterns, including heartbeat, in zebrafish embryos (Fig. 5C). PA was not cytotoxic in Vero cells at the concentrations tested (0.15 ~ 0.60 μM). Furthermore, the toxicity of PA in zebrafish embryos was assessed using embryo mortality and heartbeat disturbances. PA did not affect the survival rate of zebrafish embryos. Although weak toxicity was observed at 0.60 μM PA, it was not significant (Fig. 5D). Furthermore, the heart rate was not influenced by PA. After 4 days of PA treatment edema was observed in one zebrafish embryo (Fig. 5E and Fig. 6E).
We previously reported that secondary metabolite fractions obtained from marine bacteria collected in Jeju have excellent antioxidant, anti-inflammatory and anticancer activity (Kim et al., 2014). We showed that secondary metabolite fractions from
Preparative CPC is a non-solid support, preparative, liquid-liquid separation process that is useful for the isolation of biologically active compounds from plants and marine algae (Kim et al., 2006; Lee et al., 2013 and 2014). We successfully isolated three secondary metabolites, cyclo(L-[4-hydroxyprolinyl]-L-leucine), cyclo(L-Phe-trans-4-hydroxy-L-Pro), and PA, from
Flow cytometry revealed that PA increased the proportion of cells in the G0/G1 phase, indicating that PA suppresses HL-60 cells growth in association with cell-cycle arrest in the G0/G1 phase. Cell-cycle arrest in cancer cells is a major indicator of anticancer activity. In particular, anticancer agents may alter the regulation of the cell-cycle machinery, resulting in arrest during various phases of the cell cycle and, reduced cancer cell proliferation.
Apoptosis plays an important role in the maintenance of cellular homeostasis by regulating cell division and cell death (Yan et al., 2008). Cancer cells are characterized by uncontrolled proliferation and reduced apoptosis. Thus, activation of apoptotic pathways is a key mechanism by which cytotoxic drugs kill cancer cells (Xu et al., 2009). The Bcl-2 protein family is a critical regulator of apoptotic pathways. Many cancers, including leukemia are associated with altered expression of Bcl-2 and Bcl-xL (Young-Min et al., 2012). The anti-apoptotic protein Bcl-xL resides on the outer mitochondrial membrane and inhibits apoptosis in the presence of various apoptotic stimuli, thereby promoting cell survival (Kang and Reynolds, 2009). In contrast Bax is a pro-apoptotic Bcl-2 family protein that resides in the cytosol and translocates to the mitochondria upon apoptosis induction (Cory and Adams, 2002). PA decreased Bcl-xL expression in HL-60 cells, suggesting that decreased Bcl-xL expression may mediate PA-induced apoptosis. Additionally, caspase-3 and PARP cleavage correlated with PA-induced apoptosis in HL-60 cells. Caspase-3 is a key regulator of apoptosis, and is either partially or totally responsible for the proteolytic cleavage of many key proteins, including PARP (Fernandes-Alnemri et al., 1994). PARP is important for cell viability; however, cleavage facilitates cellular disassembly and serves as a marker of cellular apoptosis (Konopleva et al., 1999; Oliver et al., 1998). Hence, PA most likely induce apoptosis through a caspase-dependent pathway.
ROS generation in cancer cells is often increased after treatment with anticancer agents (Szatrowski and Nathan, 1991; Schumacker, 2006; Trachootham et al., 2006). Previous studies demonstrated that high ROS levels induce cellular damage (Pelicano et al., 2004; Valko et al., 2006; Li et al., 2007; Hseu et al., 2008) and may play an important role in mediating apoptosis (Garcia-Ruiz et al., 1997; Coyle and Puttfarcken, 1993). We determined that ROS are generated during PA-induced apoptosis in HL-60 cells. We evaluated intracellular ROS generation using flow cytometry to further clarify the relationship between ROS generation and apoptotic body formation, in PA-treated HL-60 cells. Our data indicate that PA-mediated apoptosis is associated with ROS generation.
PA is a clear, colorless to pale yellow liquid with a floral odor (Arctander, 1969). PA occurs in nature, with the highest quantities observed in evergreen trees (
In conclusion, this study demonstrated the biological mechanisms underlying the anti-cancer effects of PA in HL-60 cells. Our data reveal that PA induces growth inhibition and apoptosis, associated with the down-regulation of Bcl-xL, activation of caspase-3, and ROS generation. Therefore PA may be a potential anticancer drug candidate for leukemia.

![Inhibitory effects of two fractions (hexane and water fraction) (A) and compounds (cyclo[L-(4-hydroxyprolinyl)-L-leucine], the cyclo(L-Phe-trans-4-hydroxy-L-Pro) and PA) (B) from S. griseus extract against the growth in the HL-60 cells. HL-60 cells were incubated with various concentrations of two fractions for 48 h and the cell viability was examined by an MTT assay. Each value indicates that the mean ± standard error from three independent experiments.](http://oak.go.kr/repository/journal/15859/E1HKAL_2015_v18n1_35_f002.jpg)