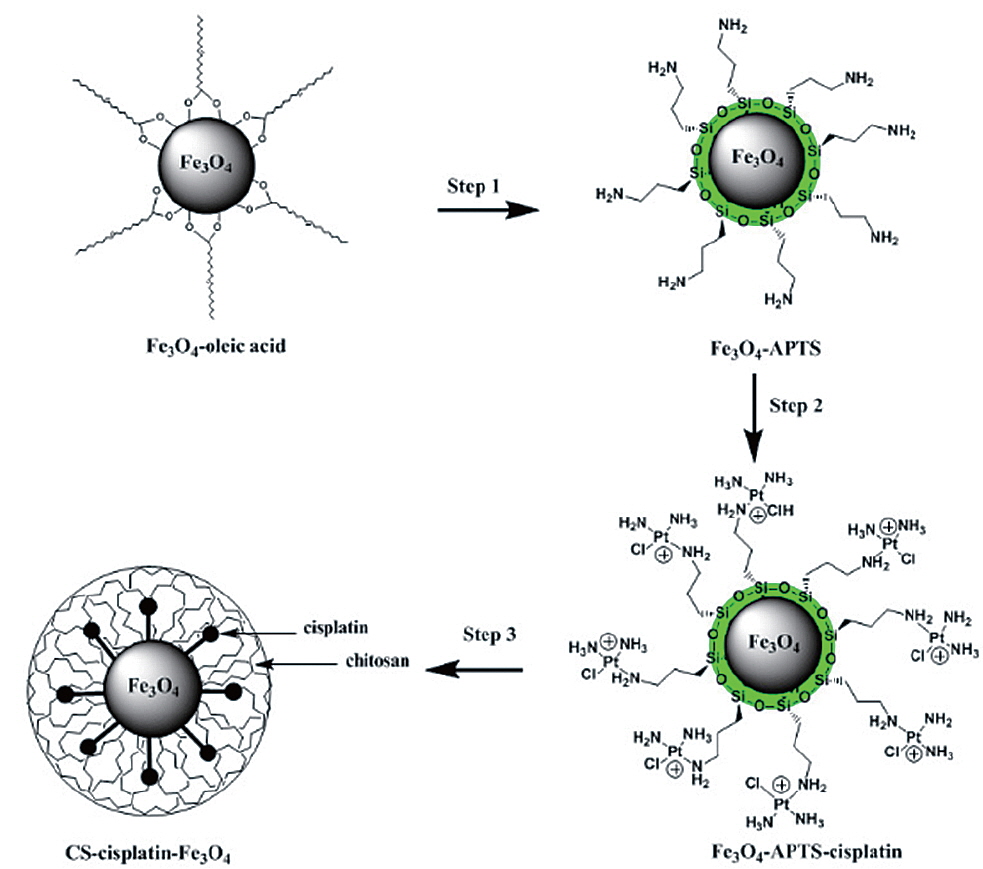
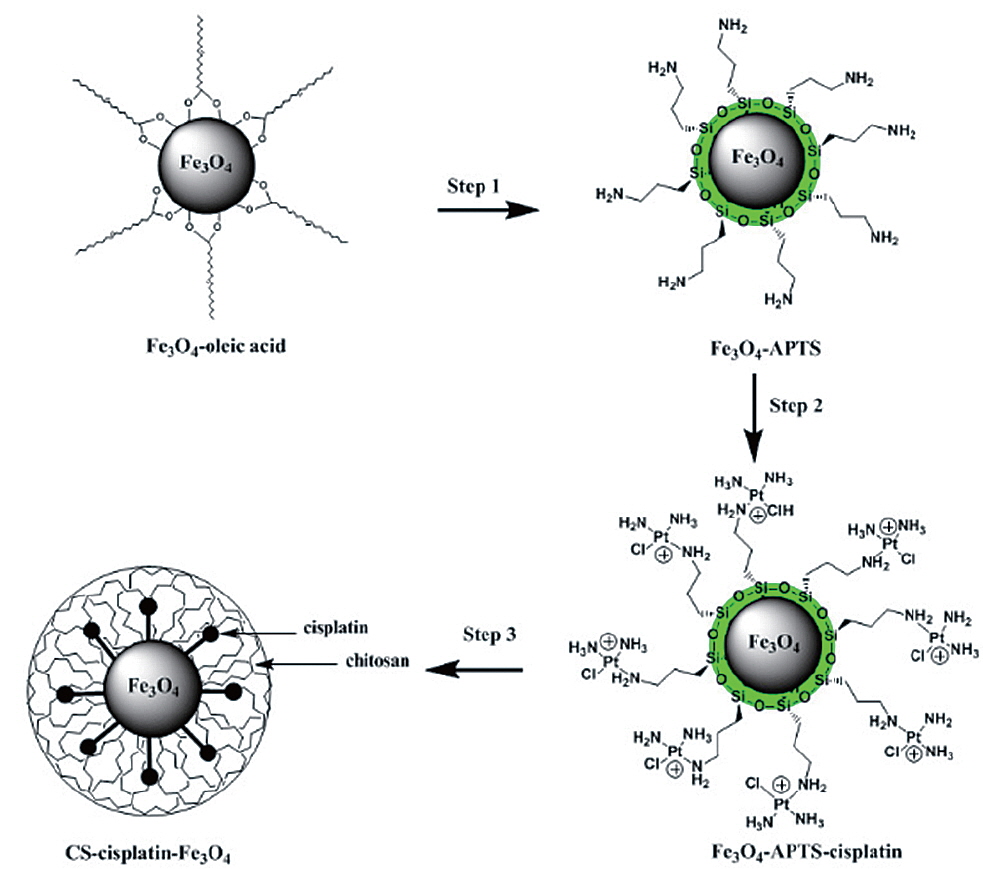
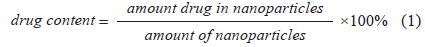A synthesis method for a chitosan-coated magnetic drug-delivery system of cisplatin is proposed. Here, cisplatin was conjugated to the surface of Magnetite (Fe3O4) nanoparticles via a (3-Aminopropyl)-trimethoxysilane (APTS) coupling agent. To reduce the cytotoxic effect of cisplatin, the magnetic drug was then encapsulated in chitosan (CS-cisplatin-Fe3O4) through the water/oil (W/O) emulsion method. The CS-cisplatin-Fe3O4 nanoparticles were synthesized in a spherical shape with a diameter of 190 nm. The cytotoxicity assay was performed using HeLa cells. The cisplatin uptake of the cells was determined using High Performance Liquid Chromatography (HPLC) to calculate the drug content. The controlled release of cisplatin was demonstrated by regulating the dissolution and diffusion of the drug through the chitosan matrix.
Natural marine materials, including agar, alginates, carrageenan, and chitin, have been of great interest to researchers for their potential application within the biomedical field due to their easy accessibility and sustainable exploitation (Kurita, 2006; Laurienzo, 2010). Among various marine biomaterials, chitosan has attracted considerable attention in light of its biodegradable and bioactive properties. Chitosan is a nontoxic, biocompatible, and biodegradable amino polysaccharide that has been used in a number of medical applications, such as aqueous solutions for drug delivery (Bhattarai et al., 2010), tissue engineering (Suh and Matthew, 2000), wound dressing (Jayakumar et al., 2011), antibacterial coatings (Jena et al., 2012), and separating membranes (Mi et al., 2001). As a natural polymer, chitosan is the deacetylated derivative of chitin, which is part of the organic matrix of exoskeletons of arthropods and endoskeletons of mollusks. Thus, it is a nontoxic biocompatible amino polysaccharide that has a number of medical applications in drug release in aqueous solutions at pH < 6.5. Specifically, chitosan oligosaccharide has been applied to a variety of magnetic nanoparticles in order to enhance their colloidal stability and circulation (Zhu et al., 2009; Bae et al., 2012).
Magnetic nanoparticles (MNPs) are currently used in various medical applications, such as drug delivery (Kohler et al., 2005; Zhang et al., 2005), magnetic resonance imaging (MRI), contrast agents (Kim et al., 2003), magneto-motive ultrasound (Oh et al., 2006), and optical imaging (Oh et al., 2007). Recently, drug-loaded Fe3O4 NPs systems have been combined with hyperthermia treatments. These methods have the added benefit that Fe3O4 NPs accumulate drugs in the targeted area and generate heat in an AC magnetic field due to hysteresis loss (Ito et al., 2004). The heating of the cancer area with Fe3O4 NPs at 42℃ can induce apoptosis of tumor cells. However, the advancement of Fe3O4 NPs for drug delivery for cancer purposes has been constrained due to the possibility of harmful effects on healthy cells. Therefore, the creating an efficient design that can facilitate controlled and targeted drug delivery to the specific regions of the cancer is a major challenge in drug delivery and development. Nanoparticle-based drug delivery and drug targeting systems are currently being developed to enhance the
Cis-diamminedichloroplatinum(II) (cisplatin), chitosan (deacetylation 85%, Mn 50 KDa, viscosity 20-300 cP), FeCl3·6H2O (98%), oleic acid (90%), 1-octadecene (90%), (3-Aminopropyl)-trimethoxysilane (APTS), tetramethylammonium hydroxide (TMAH), hydrochloric acid (37%), isopropanol, Span 80, Tween-20, and glutaraldehyde (25%) were purchased from Sigma-Aldrich Chemical Co. Sodium oleate (95%) was obtained from TCI (Korea). Petroleum ether and sodium chloride were produced by Samchun Chemicals (Korea). All chemicals were analytical grade reagents and used directly without further purification.
>
Synthesis of cisplatin-capped Fe3O4 NPs via APTS
The synthesis of Fe3O4 NPs was performed following the reported procedure (Park et al., 2004). Water-soluble Fe3O4-APTS were prepared with the two-step silanization process (De Palma et al., 2007) with little modification. APTS was used to couple cisplatin to Fe3O4 NPs. 10 mg of Fe3O4-APTS was dispersed in 10 mL of ethyl alcohol and 500 mL of TMAH 1 M (pH adjusted to 8). In the other container, 20 mg of cisplatin was dissolved in 500 mL of HCl 0.2 M and heated with a water bath at 80℃ several times. The solution was then added to a flask containing Fe3O4-APTS and stirred for 24 h. After reaction, Fe3O4-APTS-cisplatin was collected with a permanent magnet. The purification of the conjugation from the free cisplatin was performed by dialysis in 0.9% sodium chloride solution with a cellulose membrane (MWCO=7,000 Da) for three days. The Fe3O4-APTS-cisplatin was then dried under a vacuum at 85℃ for 3 h.
>
Synthesis of CS-cisplatin-Fe3O4
The typical water-in-oil (W/O) emulsion method was used to produce CS-Cisplatin-Fe3O4. 20 mg of CS was dissolved in 3 mL of acetic 1% acid. 10 mg of Fe3O4-APTS-cisplatin was dissolved in 0.5 mL of water and treated with ultrasonic waves for 10 min. These solutions were added dropwise into 60 mL of paraffin oil containing emulsion stabilizer 5% Span 80 and 0.5% Tween-20 under a stirring rate of 1,200 rpm at room temperature. After 24 h, the suspension was added with 2 mL glutaraldehyde 25% and stabilized for 3 h. The CS-Cisplatin-Fe3O4 was then collected by applying a permanent magnet, washed three times using petroleum ether, and recovered by centrifugation at 10,000 rpm. The final product was washed with isopropanol and dried under a vacuum at 70℃ for 12 h.
The crystalline structure of the Fe3O4 NPs was examined with XRD (Philips, X’Pert-MPD System, Netherlands); the changes in the chemical groups present in the nanoparticles were monitored using a FTIR spectrometer (Jasco FT/IR 4100, USA); the attachment of cisplatin to APTS-Fe3O4 NPs was detected with XPS (Thermo VG Scientific, MultiLab2000, England); the particle size was measured using TEM (Jeol, JEM- 2010, Japan); the particle shapes were observed with SEM (Hitachi, S-2400, Japan); the zeta potential was determined using a Electrophoretic Light Scattering Spectrophotometer (Otsuka Electronics, ELS-8000, Japan); and the magnetic properties were measured using SQUID (Quantum Design, MPMS XL 7.0, USA).
>
Drug content in nanoparticles
Fe3O4-APTS-cisplatin and CS-cisplatin-Fe3O4 NPs were dissolved in nitric acid (pH 3.5) at a concentration of 100 μg/mL. Subsequently, the samples were sonicated for 60 min and centrifuged at 8,000 rpm for 10 min. The supernatants were measured using HPLC. The cisplatin standard solution was prepared in 0.9% NaCl solution and used to prepare the calibration curves ranging from 1 to 10 μg/mL. The drug content was calculated using the following Eq. 1:
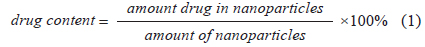
A Flexar HPLC system (PerkinElmer, USA) was used for the liquid chromatography system. A 20-μL sample was injected into the C18 column, 150 × 4.6 mm, 5 μm particle size (Agilent Eclipse XDB, USA). The mobile phase was a mixture of water/methanol/acetonitrile (50:25:25), and the detection was performed at 254 nm with a running time of 20 min.
The HeLa cells (ATCC CCL-2) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Scientific, USA) and supplemented with 10% fetal bovine serum (FBS, Thermo Scientific). The cells were incubated at 37℃ in a humidified atmosphere of 5% CO2 and 95% air, and the growth medium was changed twice a week. The cell seeding was performed by removing the medium and adding 2 mL of Trypsin (0.25%)-EDTA (0.53 mM) (Thermo Scientific) to the flask, then observing the cells under a microscope until the cell layer was dispersed. Finally, a 6-ml medium was added, and the cultures were incubated at 37℃.
The cytotoxicity of the CS-cisplatin-MNPs was measured using the WST-1 assay (ITSBio, Korea). The HeLa cells were seeded at a density of 1.0 × 105 cells/well plates and incubated for 24 h. Fe3O4-APTS, Fe3O4-APTS-cisplatin, chitosan-glutaraldehyde, CS-cisplatin-Fe3O4, and cisplatin (each with a concentration of 50 mg/mL) were added to the culture media in the plate, and the untreated cells were used as controls. The cells were incubated in humidified 5% CO2 at 37℃ for 12, 24, or 48 h, respectively. At the end of the incubation time, 10-μL WST-1 solution was added to each well plate. After 4 h of incubation, the plates were shaken for 1 m, and the absorbance peak at 450 nm was measured and calculated with Eq. 2 using a microtiter plate reader (Molecular Device, USA).

>
In vitro release of cisplatin and drug uptake
The amount of cisplatin released and the uptake into the HeLa cells was determined using HPLC, as reported earlier (Lopez-Flores et al., 2005). The HeLa cells (1.0 × 106 cells/well) were treated with CS-cisplatin-Fe3O4 with a final concentration of 50 μg/mL and were incubated for 3, 6, or 12 h in a 5% CO2 atmosphere at 37℃. The untreated cells samples were incubated for the same period. After incubation, the cells were washed with PBS and harvested with 500 μL of trypsin containing EDTA. Subsequently, they were lysed with buffer (Tris 100 mM, EDTA 5 mM, NaCl 200 mM, 0.2% SDS at pH 8) for three hours at 55℃. The homogenate was filtered and extracted with 80 μL of chloroform by vortexing at the maximum speed for one minute on a Vortex-Genie (Scientific Industries, USA), and then separated by centrifugation at 10,000 rpm for 5 min. Finally, 20 μL of the chloroform layer was injected into the HPLC system under the same chromatographic conditions.
>
Characterization of Fe3O4 NPs
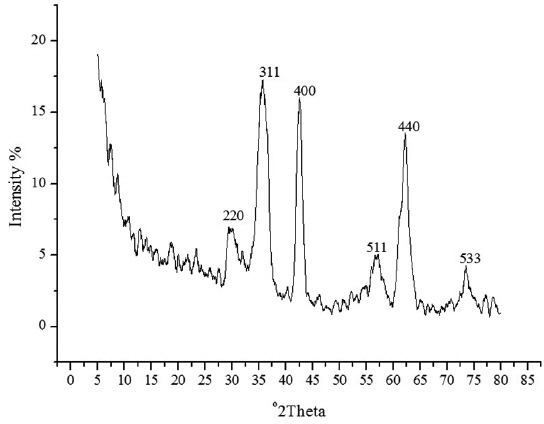
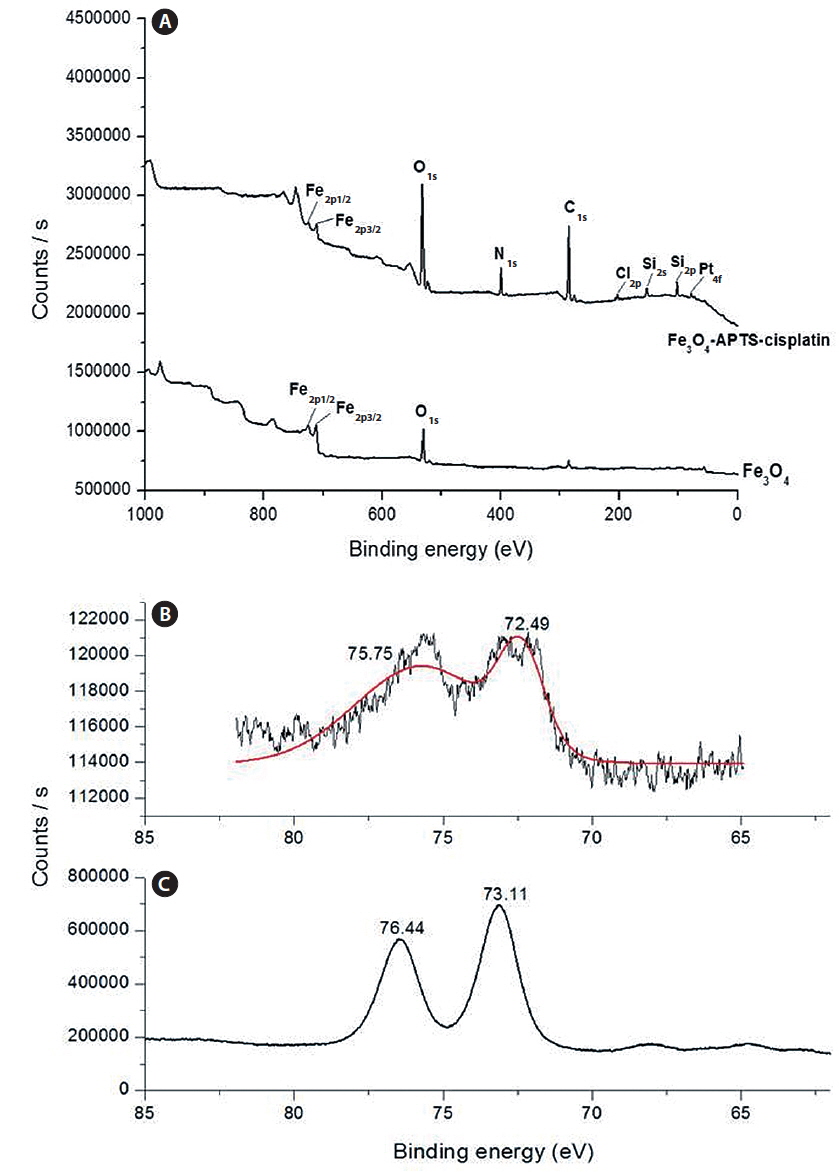
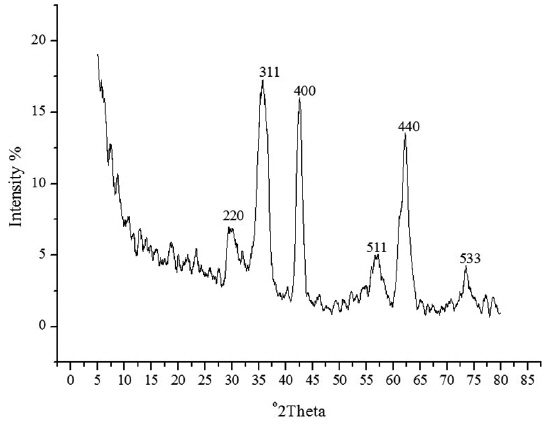
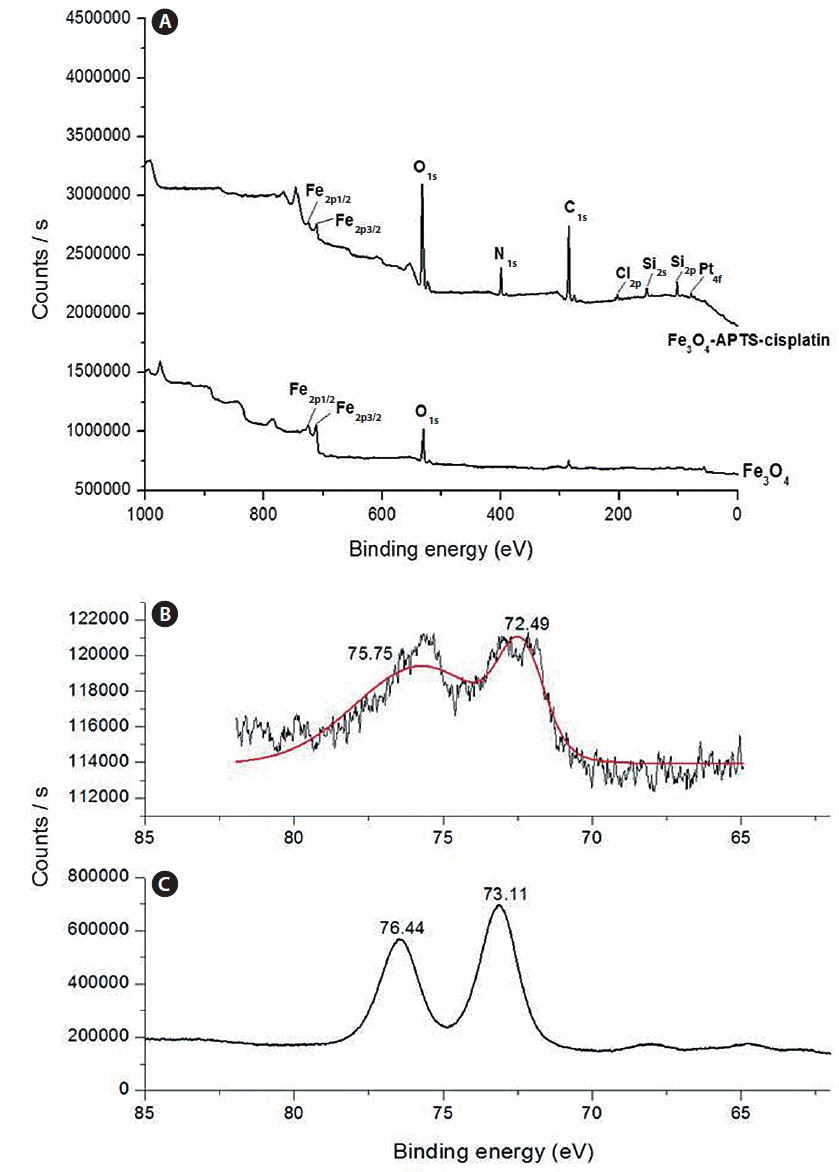
The spherical monodisperse iron oxide nanoparticle was prepared through thermal decomposition of Fe-oleate in the presence of oleic acid as a surfactant. Fig. 2 represents the XRD patterns of Fe3O4 at 2θ = 30.3, 35.5, 36.5, 42.5, 57.0, 62.2, and 73.4, corresponding to 220, 311, 400, 511, 440, and 533 Bragg reflection planes, which were observed in all of the samples. These peaks are consistent with the standard pattern of Fe3O4 (JCPDS no. 79-0418) with a cubic inverse spinel structure. The results indicated that the crystal structure of Fe3O4 NPs was not changed during the modification with oleic acid. The mean crystal sizes were determined by the Debye–Scherrer equation with XRD data, D = Kλ/(βcosθ), where K is a constant (K = 0.94 for Cu-Kα), λ is the wavelength (0.15405 nm for Cu-Kα), β is the peak width of the half-maximum, and θ is the diffraction angle (Sun et al., 2009). Thus, the crystal sizes of the Fe3O4 NPs were found to be approximately 14.25 nm. To confirm the relative surface composition of the functionalized Fe3O4 NPs, XPS was used, as shown in Fig. 3a. The binding energy of Fe2p3/2 and Fe2p1/2 were 711.1 eV and 724.5 eV, respectively, which are close to the standard data of Fe3O4.
>
Characterization of Fe3O4-APTS-cisplatin
The presence of oleic acid on the surface of Fe3O4 NPs from the above result caused the particles to become hydrophobic. APTS functionalized the Fe3O4 NPs to modify the particle to become hydrophilic. As previously established, silane can be applied to exchange the original hydrophobic oleic acid ligand from the surface of the Fe3O4 NPs and increase biocompatibility (Jana et al., 2007). The influence of the end group of the APTS coating gave the nanoparticles a water-dispersible effect. In the next step, APTS was used to couple cisplatin to Fe3O4 NPs through a nucleophilic substitution reaction between the chloride ligand and the amino group of the silane. The XPS data of Fe3O4-APTS-cisplatin is shown in Fig. 3a. The binding energy of Si2s and Si2p were found as 156.4 and 105.4 eV, respectively, which are similar to the previous data of silane (Wu and Xu, 2005). The peaks of C1s at 290.9 eV are close to the standard data of alkyl chains, signifying it derived from the alkyl chain of APTS. The binding energy of N1s was 404.0 eV, which is close to the standard data of an amine group. The presence of cisplatin on the surface of the MNPs is shown through the peak of Pt4f at 78.6 eV and Cl2p at 201.7 eV. In order to verify the bond between APTS and cisplatin, the platinum peak was compared with the Pt4f peak of the cisplatin standard solution. The results showed there were shifts in the position of the peaks to lower energy values of 73.11 and 76.44 eV for the cisplatin standard solution (Fig. 3c) and to 72.49 and 75.75 eV for the Fe3O4-APTS-cisplatin (Fig. 3b). These lower energies indicated a change in the binding state of the platinum. As previously reported, the particles become less polar as many platinum-bound chlorines could be substituted by the terminal amine group of APTS (Bhowmick et al., 2010).
>
Characterization of CS-cisplatin-Fe3O4
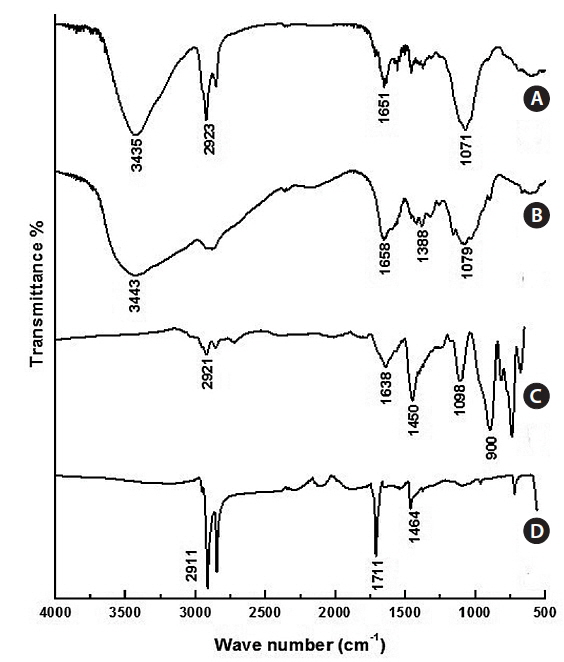
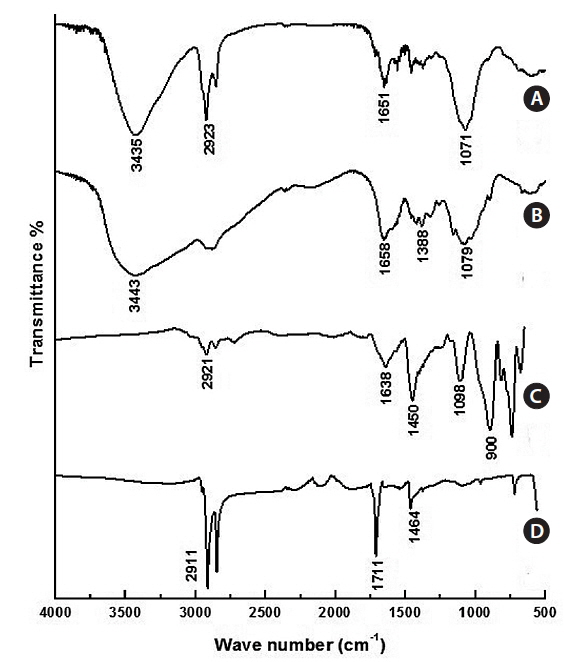
The typical FT-IR spectra of Fe3O4 NPs are shown in Fig. 4a. The C=O at 1,711 cm-1, the C-H stretch at 2,911 cm-1, and the CH2/CH3 bending at 1,464 cm-1 are evidence of oleic acidcoated Fe3O4 NPs. Fig. 4b shows the FT-IR of Fe3O4-APTS. The peaks at around 1,638 cm-1 were ascribed to the –NH2 terminal of APTS, and the Si-O-Si bridges were found at 1,098 cm-1. The absence of oleic acid peaks and appearance of silane peaks in the Fe3O4 NPs indicate that the ligand exchange was performed successfully. The FT-IR peaks of CS-cisplatin-Fe3O4 (Fig. 4d) were compared with those of pure chitosan (Fig. 4c). The same pattern was shown for the O-H region in 3,435cm-1 and the C-O stretching at 1,071 cm-1. In the case of CS-cisplatin-Fe3O4 (Fig. 4d), a sharp peak was observed at 1,651 cm-1 that derived from the glutaraldehyde imine group (C=N). The reaction between chitosan and glutaraldehyde made the amino group of chitosan convert into an imine group (C=N). The strong peak of the C-H stretch at 2,923 cm-1 resulted from the alkane groups of APTS.
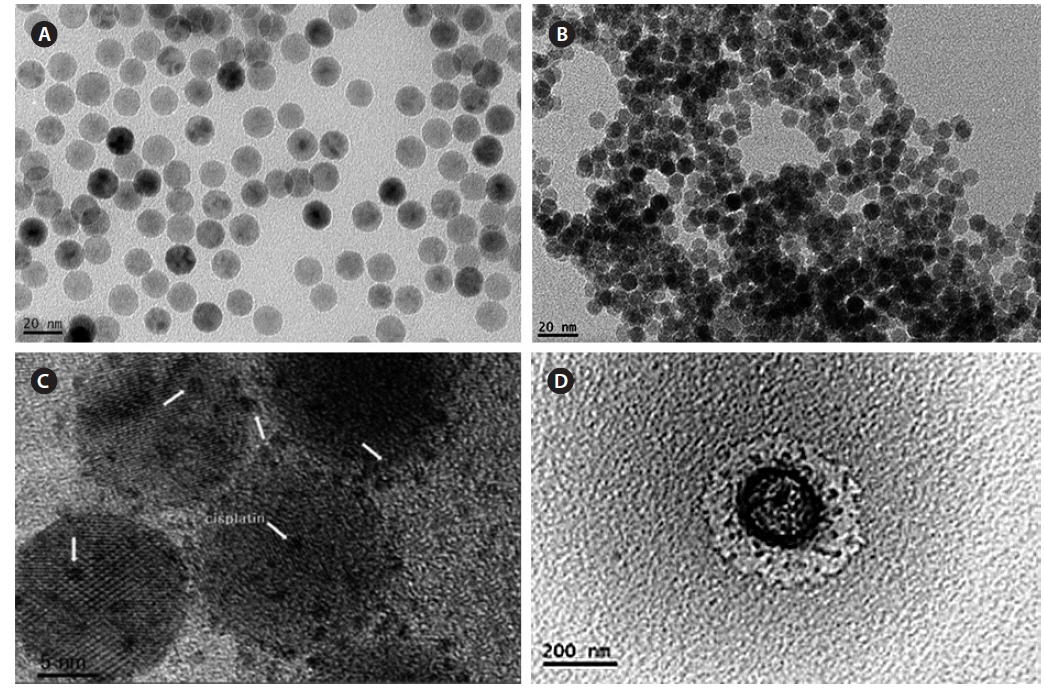
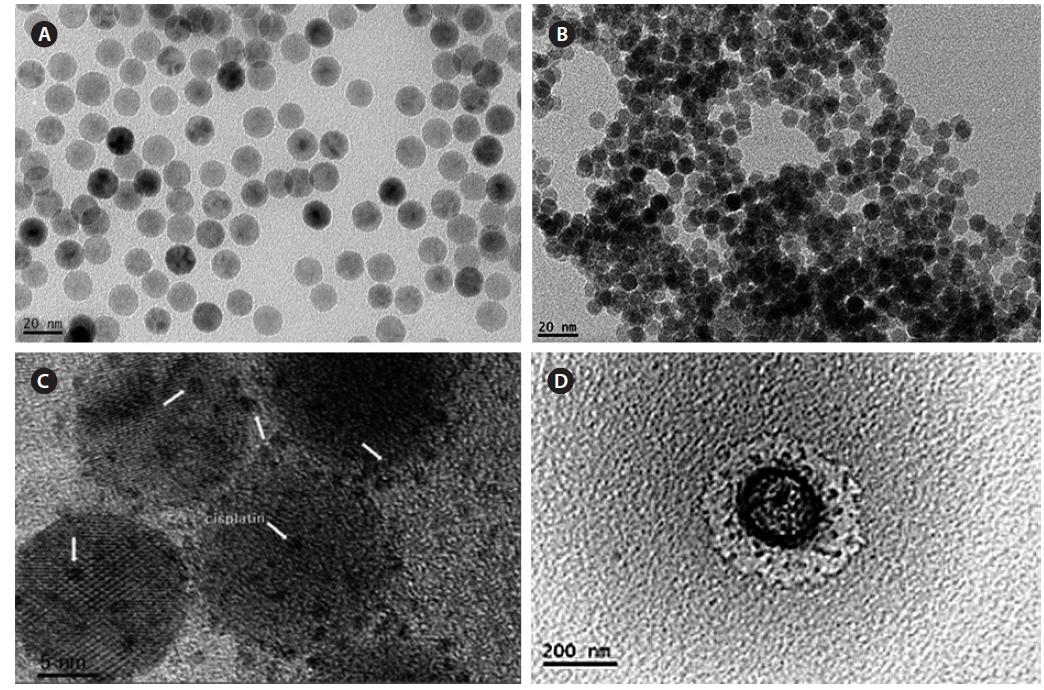
Fig. 5 shows a TEM image of all processes of magnetic nanoparticles, and the size was measured by ImageJ software. Fig. 5a shows Fe3O4-oleic acid, and the diameter was 14.5 nm, which is close to the calculation using the Debye–Scherrer equation in our XRD data. Fig. 5b shows the Fe3O4-APTS nanoparticles were spherical and 15.87 nm in size, and they did not appear to aggregate in water. Cisplatin-capped Fe3O4 via an APTS coupling agent with an average size of 8.8 nm can be seen in Fig. 5c. Dark spots at the Fe3O4 NPs surface were indicative of cisplatin. Furthermore, the TEM image of CS-cisplatin Fe3O4 (Fig. 5d) shows the effect of glutaraldehyde crosslinking amine functional groups of chitosan, which had a spherical shape and diameter of 190 nm.
>
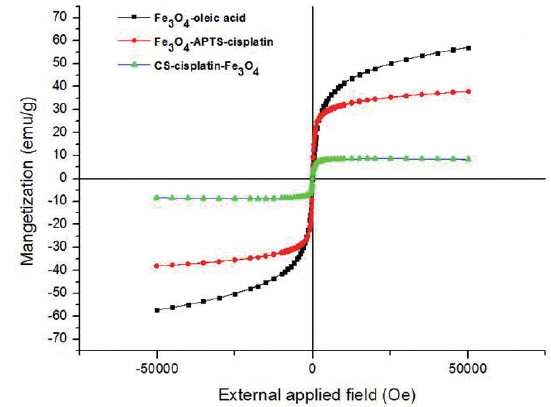
Magnetic properties measured by SQUID
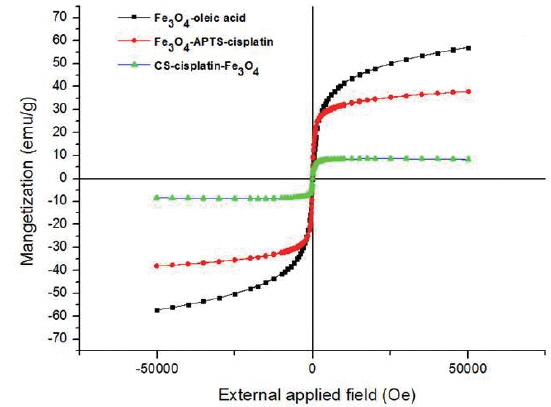
Fig. 6 shows the magnetic curves as a function of applied field at 300K obtained for dry powders of Fe3O4-oleic acid, Fe3O4-APTS-cisplatin, and CS-cisplatin-Fe3O4, respectively. The magnetization saturations were found to be 57.2 emu/g for Fe3O4-oleic acid, 38.0 emu/g for Fe3O4-APTS-cisplatin, and 8.5 emu/g for CS-cisplatin-Fe3O4. The magnetization value decreased after coating due to the existence of silane and chitosan, which formed polymerized multilayers. In addition, no hysteresis was found in any of the samples, indicating that these nanoparticles have superparamagnetic properties.
>
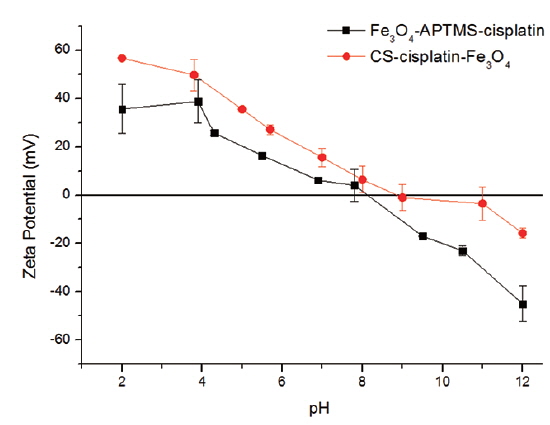
Drug content and zeta potential
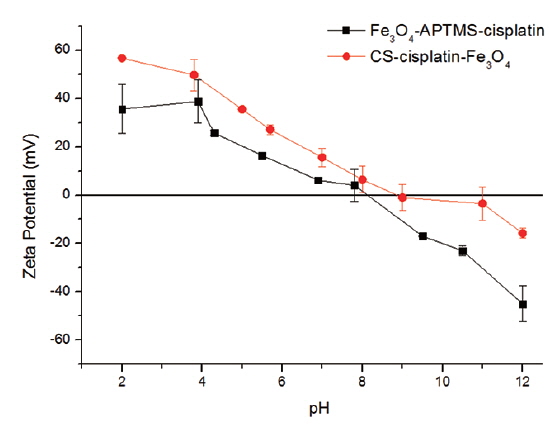
The percentages of cisplatin content (m/v) in the nanoparticles are shown in Table 1. Cisplatin content in Fe3O4-APTS was 27%, while in the Fe3O4-CS system, it was 18%. The amount of cisplatin seems to be influenced by the amount of iron encapsulated by chitosan. The measured zeta potential values of these particles are shown in Fig. 7.
[Table 1.] Drug content of Fe3O4-APTS-cisplatin and CS-cisplatin-Fe3O4

Drug content of Fe3O4-APTS-cisplatin and CS-cisplatin-Fe3O4
It is shown that Fe3O4-APTS-cisplatin and CS-cisplatin-Fe3O4 were both positively charged at acidic pH, which confirms the presence of amino groups on the particles in their protonated form and thus confirms the presence of chitosan on the particle surface. In the case of CS-cisplatin-Fe3O4, it was observed that the surface potential indicated precipitation starting from pH 6.0 and decreased to 0 at around pH 9, indicating the amino groups were deprotonated. However, the pKa of chitosan was 6.5, and when the pH increased, chitosan lost its protonated amino group, promoting intramolecular hydrogen bonding and causing the chitosan to precipitate.
>
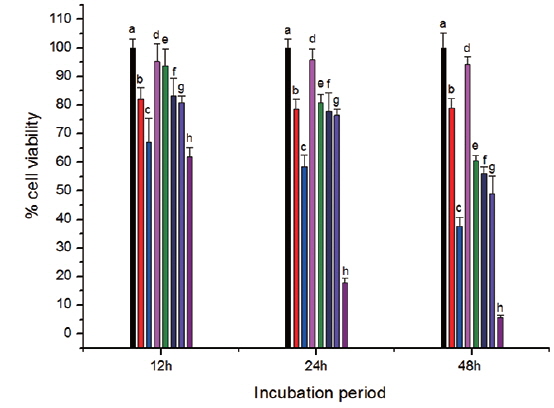
In vitro cell viability assay
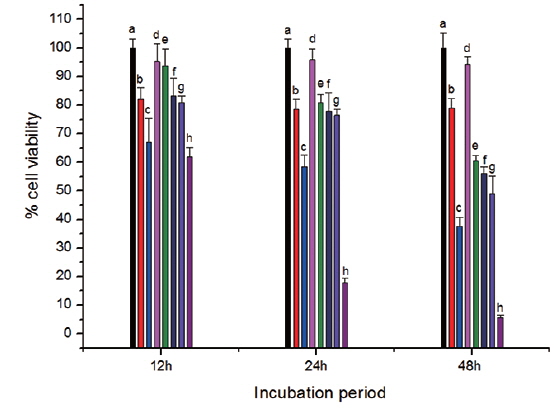
The toxicity of CS-cisplatin-Fe3O4 was demonstrated by a WST-1 cytotoxicity assay. This assay is based on the cleavage of the tetrazolium salt to water-soluble formazan by the succinate-tetrazolium reductase system, which belongs to the respiratory chain and is only in the viable cells (Ishiyama et al., 1996). Therefore, the amount of formazan dye is directly related to the number of living cells. Cell viability was observed after cells were incubated for various time periods: 12, 24, and 48 h. Fig. 8 shows that cell viability decreased more rapidly for cisplatin and Fe3O4-APTS-cisplatin. As confirmed by previous literature (Jamieson and Lippard, 1999), cisplatin can promote abnormal linkages in DNA through chloride ligand crosslinking and hydrogen bonding of amine ligands. In the case of Fe3O4-APTS-cisplatin, the cytotoxic action came from the one chloride ligand remaining and the hydrogen-bonding interaction of the NH2R group to the oxygen-phosphate of DNA (Komeda et al., 2011). The cell viability of Fe3O4-APTS and chitosan appeared stable for all incubation periods. Neither had a significant effect on cell viability, and both were categorized as non-toxic. For CS-cisplatin-Fe3O4, viability declined during incubation times, indicating that cisplatin was continually released. However, the glutaraldehyde crosslink chitosan microsphere demonstrated minimal cisplatin toxicity.
>
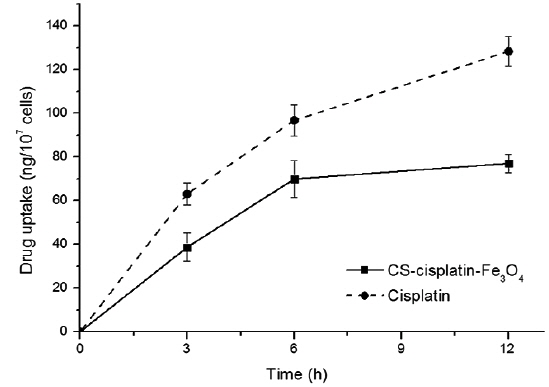
In vitro release of cisplatin
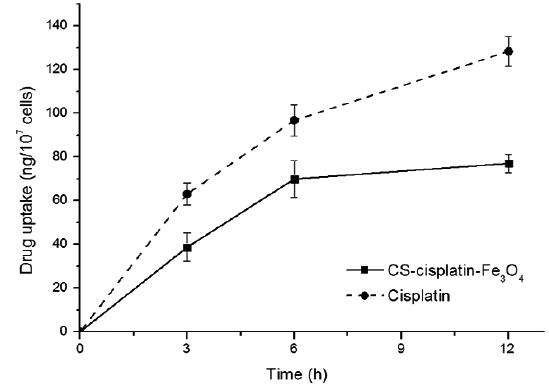
The intracellular platinum concentrations increased continuously as a consequence of DNA-platination. In order to demonstrate the effect of chitosan on controlling drug release, we compared the cell-uptake behavior to a cisplatin drug with the same concentration of cisplatin content in its nanoparticles. Fig. 9 indicates that cisplatin accumulation within HeLa cells was more accelerated. The drug-release rate from the glutaraldehyde crosslinked chitosan microsphere was controlled by the dissolution and diffusion of the drug from the chitosan matrix (Wang et al., 1996).
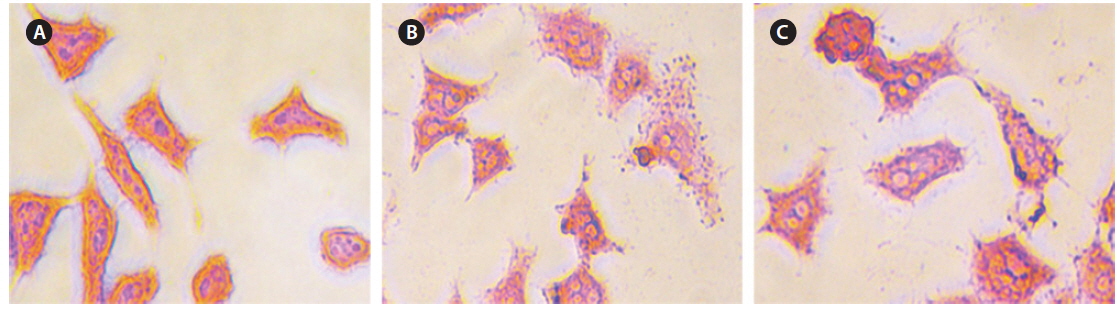
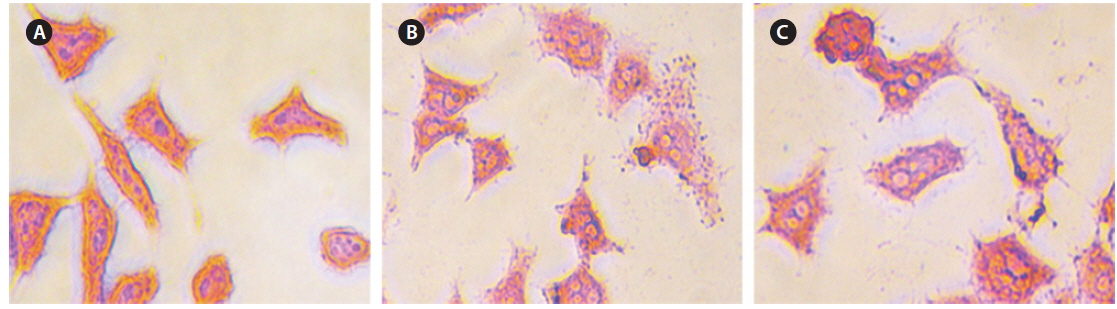
Intracellular Prussian blue staining was used to demonstrate the presence of ferric iron in the cytological preparations of HeLa cell suspensions after 48 h incubation. Fig. 10 shows obvious visual evidence of the presence of iron in panels (b) and (c) compared with untreated cells (a), and the amount of iron was increased for higher concentrations of CS-cisplatin-Fe3O4 MNPs.
The present work proposed a method for developing a magnetic drug-delivery system for cisplatin intended to be used in specifically targeted cancer therapy. In our synthesis procedure, we kept the morphology of the nanoparticles in a spherical shape. For biomedical applications, it is crucial that magnetic nanoparticles are spherical in shape and uniform in size. The spherical shape enables the uniform immobilization of bioactive molecules onto the surfaces of the particles, and the uniform size endows the particles with uniform magnetic properties. We used chitosan encapsulation to optimize the pharmaceutical action while reducing the cytotoxicity of cisplatin. In addition, to prepare a slow-release system, we controlled cisplatin from the chitosan microsphere, which can be observed in the intracellular concentration–time profiles (Fig. 9), and chitosan was shown to reduce the release rate of cisplatin. In conclusion, our results support the idea that chitosan nanoparticles are a suitable carrier for controlled release and a magnetic drug-delivery system using cisplatin and that they should be promoted for further gene-based therapy and magnetic hyperthermia.