Investigation of nanoscale and molecular scale requires different approaches than that of bulk. The current widespread application of nanomaterials and their molecular limits is increasing interest in discovering the exact molecular species that can be formed at such scales. One of these structures is diamondoids used to investigate the molecular and nanoscale limits of cubic diamond structure materials [1]. Carbon diamondoids were first found in petroleum and were synthesized and purified in the laboratory afterwards. Diamondoids are beneficial in understanding how these blocks combine to form nanoscale and bulk. They have been used in crucial applications in areas such as medicine and industry [2]. Diamondoid structures are not only limited to diamond but have been extended theoretically to other IV elements or IV-IV compounds, such as Si, Ge, Sn, and GeSi [3,4], from which Si has been prepared in the laboratory [5]. The zincblende crystal structure is a diamond analogue structure for III-V, II-VI, and I-VII compounds between two different elements, such as BN, ZnO, and CuI. The application of diamondoid structures to III-V compounds, such as GaAs and InAs, revealed important information concerning the electronic and spectroscopic properties of these possible structures at the nanoscale [6,7].
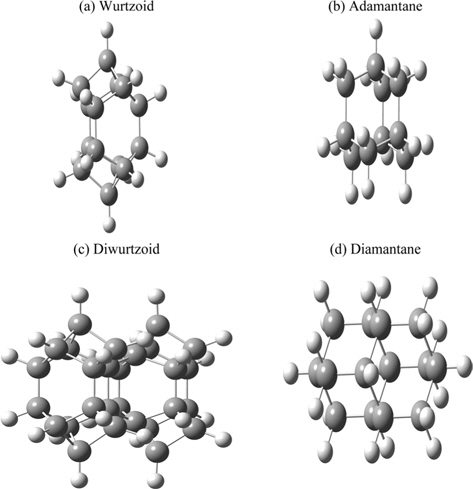
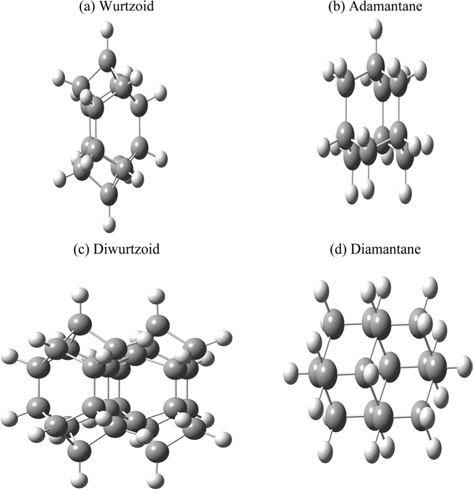
Hexagonal solid-state structures (including wurtzite) are more abundant than diamond and zincblende structures. This is obvious from the number of compounds and studies found in the literature. A hexagonal diamond called lonsdaleite can be considered to be made up of interlocking rings of six carbon atoms in a boat conformation instead of the chair conformation of cubic diamond [8]. In the present work, we propose a molecule (C14H14) that can be used as building blocks of hexagonal diamond-type crystals and nanocrystals including wurtzite structures. This molecule and its combined blocks are similar to diamondoid molecules that are used as building blocks of diamond. The hexagonal part of this molecule is included in the C12 central part of this molecule [8]. The hexagonal part can be repeated to increase the ratio of hexagonal to diamond and other structures, as will be discussed below.
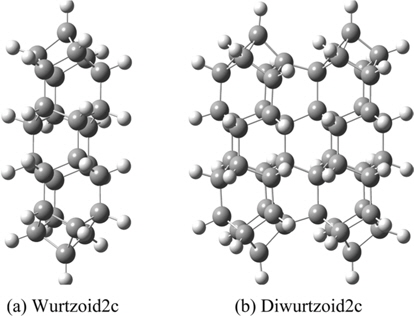
The all-electron density functional theory (DFT) at the level of local spin density approximation (LSDA) with valence triple-zeta 6-311G(d,p) basis that includes hydrogen polarization functions is used. Calculations include ethane (C2H6) and cyclohexane (C6H12) to show variation of different properties investigated in the present work. Diamondoids are named with reference to the number of their building blocks. The smallest diamondoid is adamantane (C10H16). Other investigated diamondoids in the present work include diamantane (C14H20), trimantane (C18H24), tetramantane (C22H28), hexamantane (C26H30), octamantane (Ge40H42) and dodecamantane (C52H50). The last two molecules can be considered nanocrystals since at least one of their dimensions is greater than 1 nm. The proposed molecule that carries a hexagonal signature (C14H14) is called “wurtzoid.” Combining wurtzoids together will result in diwurtzoid (C28H24), triwurtzoid (C42H30), etc. As we explained in the introduction, the hexagonal part of this molecule is included in the C12 central part of the molecule (Fig. 1). Repeating this part will produce other molecules that have higher hexagonal ratios, such as the molecule C26H26 that we call wurtzoid2c. This molecule has twice the height of the conventional hexagonal diamond unit cell. This is similar to the various hexagonal cells encoded as 2H, 4H, 6H, etc. Fig. 1 compares diamondoids and the proposed wurtzoids. Fig. 2 shows the second type of wurtzoids (wurtzoid2cs). The second type of molecules can be also combined to form diwurtzoid2c (C52H44), triwurtzoid2c (C78H54)...etc. The c axis length of wurtzoid2c and its combinations exceeds the 1 nm threshold of the nanoscale region. The wurtzite phase that has 100% hexagonality should be reached as ∞wurtzoid∞c, in which the number of surface C and H capping atoms (similar to capped (3, 0) carbon nanotubes) become negligible in relation to the overall number of atoms. Finite multiples of wurtzoid3c (C38H38) are practically sufficient to explain the trend of nanoscale hexagonal diamond properties as discussed below.
The vibrational infrared (IR), nuclear magnetic resonance (NMR), and ultraviolet-visible (UV-Vis) spectra of the proposed diamondoid and wurtzoid molecules and nanocrystals are also calculated. The frequencies of IR vibrations are corrected using the 0.988 scale factor that is usually used with the theory and basis used in this study (LSDA/6-311G(d,p)) [9]. The optimized geometries are also used to calculate the 1H-NMR spectra. The UV-Vis is calculated using excited-state energy calculation, including up to 40 states, by the time-dependent DFT (TD-DFT) method. Natural bond orbital (NBO) population analysis is performed to study the charge transfer and the kinds of hybridized molecular orbitals formed in the proposed wurtzoids and diamondoids. All the calculations are performed using the Gaussian 09 program [10]. Geometrical optimization calculations in this program restrict the maximum force on any atom to be less than 0.00045 Hartrees/Bohr. The root mean square (RMS) force of all atoms should be less than 0.0003 Hartrees/Bohr. The maximum displacement of any atom should be less than 0.0018 Å. The RMS displacement of all atoms should be less than 0.0012 Å.
The stoichiometry of the diamondoids and wurtzoids presented in the previous section demonstrates that the ratio of hydrogen atoms to carbon atoms is greater in diamondoids than in wurtzoids. This fact leads to many differences in the structural and electronic properties of these molecules as seen in following figures.
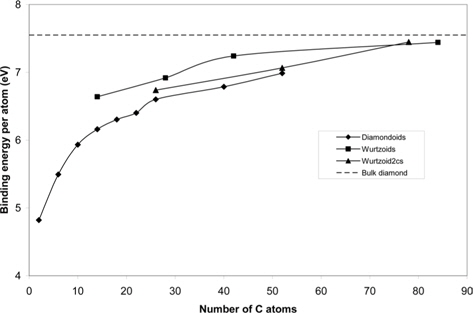
Fig. 3 shows the binding energy per atom of diamondoids, wurtzoids, and wurtzoid2cs. The results are compared with the binding energy of bulk diamond at 7.55 eV [11]. We can conclude from this figure that wurtzoids are tighter structures than diamondoids. This might be attributed to the compensation of C-H bonds with C-C stronger bonds evident from the stoichiometry of the two structures.
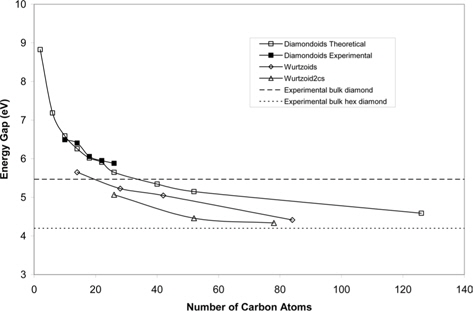
Fig. 4 shows the energy gap of diamondoids and wurtzoids as a function of the number of carbon atoms. It can be seen that it follows the quantum confinement effect of decreasing gap as a function of the number of carbon atoms. The experimental bulk diamond and hexagonal diamond gaps are 5.47 and 4.2 eV, respectively [1,12]. These limits are shown in Fig. 4. Fig. 4 also shows that, with the exception of adamantine, the present theory underestimates the energy gap of diamondoids and bulk diamond [13]. It seems a good future practice to look for different methods, such as DFT+U. The energy gap of diamondoids is far below that of bulk diamond [1]. This can be explained as follows. The electron affinity of carbon atoms is greater than that of hydrogen atoms. As a result, some extra electronic cloud is transferred to the inner core of diamondoids, which reduces the value of the energy gap below that of bulk diamond. As the cluster of atoms increases in size, this effect diminishes and the energy gap increases to reach the bulk value. In addition to the above explanation, the DFT method is known to underestimate the energy gap [1]. It is important to note that, in our calculations, the trend of the energy gaps of adamantane, wurtzoid, wurtzoid2c, and wurtzoid3c (the last one is not shown in Fig. 4) is 6.59, 5.65, 5.07, and 4.8 eV respectively. These values are consistent with the trend of bulk diamond and hexagonal diamond gaps mentioned earlier. The results obtained using the present theory seems to match better with wurtzoids than diamondoids. The higher symmetry of wurtzoid surfaces (all atoms at wurtzoids surfaces are bonded to one hydrogen atom) in comparison to diamondoids (some atoms at diamondoids surface are bonded to one, and others are bonded to two hydrogen atoms) might be the explanation. Symmetry is an important factor in quantum calculations, which is evident in the shapes of orbitals themselves.
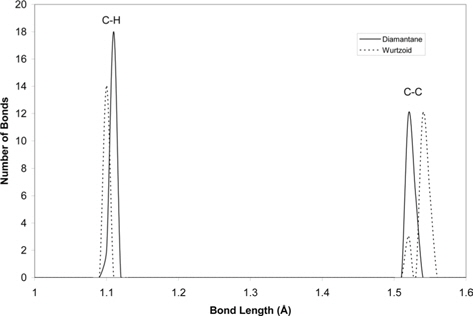
Fig. 5 compares the distributions of bond lengths of wurtzoid and diamantane molecules. We chose these two molecules because they contain the same number of carbon atoms (14 atoms). Only two kinds of bonds exist in cubic or hexagonal diamond, namely, C-H and C-C. The number of C-H bonds in diamantane is higher than in wurtzoid because it contains a higher number of hydrogen atoms. The C-H bond length of wurtzoid is slightly smaller than that of diamantane. This is because some carbon atoms in diamantane are connected to two hydrogen atoms. This phenomenon is well known and documented for carbon and other group IV elements that are connected to more than one hydrogen surface atom [14]. The truncated number of hydrogen atoms in wurtzoid relative to diamantane is compensated by increasing the number of C-C bonds. The electronic charges that are transferred from hydrogen atoms are used to reduce the length of C-C bonds in diamantane. This is not the case in longer wurtzoid bonds because it has a smaller number of hydrogen atoms. As a result, the high peak in wurtzoid indicates longer C-C bonds. The smaller C-C peak for wurtzoid is due to the vertical C-C bonds shown in Fig. 1a. This peak is an indication of the hexagonal part of wurtzoid. This small peak increases as diwurtzoid, wurtzoid2c, wurtzoid3c etc.
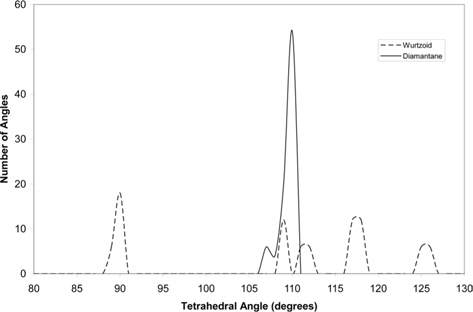
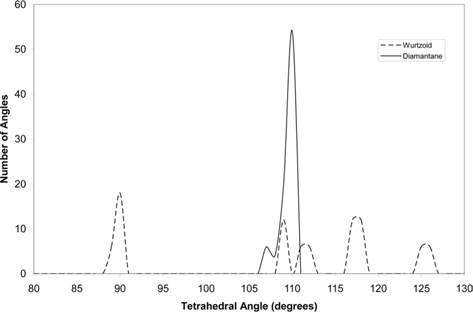
Fig. 6 compares the tetrahedral angles of wurtzoid and diamantane. In diamantane, all the angles are around the ideal tetra-hedral angle of diamond structure 109.47º [15]. The two tallest wurtzoid peak angles are nearer to the ideal hexagonal angles near 90º and 120º. Some of the angles are also near the 109.47º value of cubic diamond, indicating partial hexagonality due to the effect of capping C atoms (other than the C12 central part) and hydrogen surface atoms.
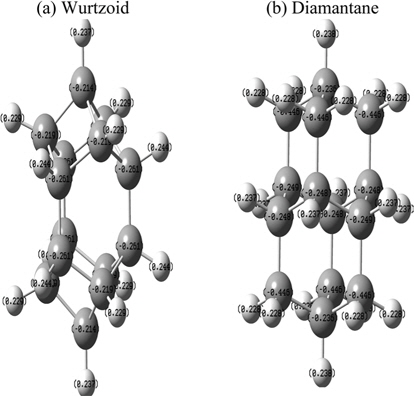
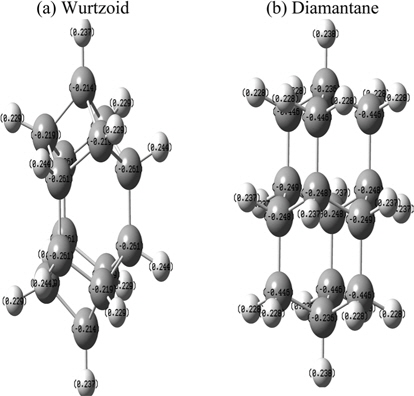
Fig. 7 shows the atomic charges of wurtzoid and diamantane obtained using NBO population analysis. The H approximate average atomic charges are 0.23 au for both diamantane and wurtzoid. Because of the higher number of hydrogen atoms in diamantane, C atoms that are connected to two H atoms are overcharged with −0.45 au, while the other C atoms have (on average) a negative H charge (−0.23 au). As a result, diamantane is more strongly affected by charge transfer than wurtzoid. This is the case for all other diamondoids since the number of H atoms in diamondoids is always larger than that in wurtzoids with the same number of carbon atoms. These charges have intense effects on the physical properties of both molecules. Some of these effects were discussed above, such as energy gap and bond length. Other effects will be discussed below. The bonding for C valence orbitals is in the ranges of (2s0.982p3.213p0.02)-(2s0.942p3.313p0.02) for wurtzoid and (2s0.932p3.293p0.01)-(2s0.992p3.443p0.01) for diamantane. These values differ from the ideal sp3 bonding because of the absorbed surface H electronic charges. They also show that absorbed H electronic charges accumulate mainly on valance p orbitals and not s orbitals. The ranges of charges in the C valence orbitals of wurtzoid are 4.21-4.27 au and 4.23-4.44 for diamantane (normally 4 electrons are in C valence orbitals), which explains the reduction of energy gap below that of bulk diamond for higher diamondoids and wurtzoids. It also explains the differences in many other properties between wurtzoids and diamondoids as discussed below.
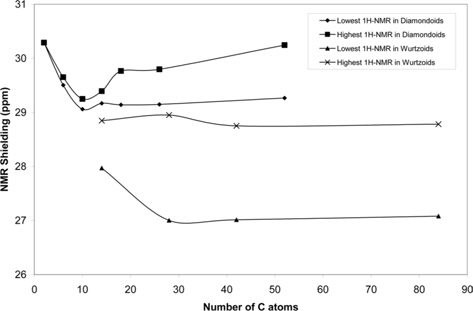
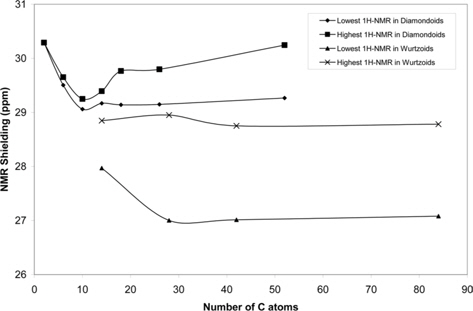
Fig. 8 shows the variation of the 1H-NMR spectra of diamondoids and wurtzoids as a function of the number of C atoms. The minimum and maximum NMR shieldings are shown. Other shielding lines are between these two lines. As discussed above, the atomic charges of diamondoids are higher than those of wurtzoids,which results in a higher effect of an external magnetic field, an electric field, and electromagnetic radiation. As expected the 1H-NMR shielding spectra of diamondoids are higher than those of wurtzoids. This is also the case of small ethane and cyclohexane molecules that have a higher (number of H atoms)/(number of C atoms) ratio.
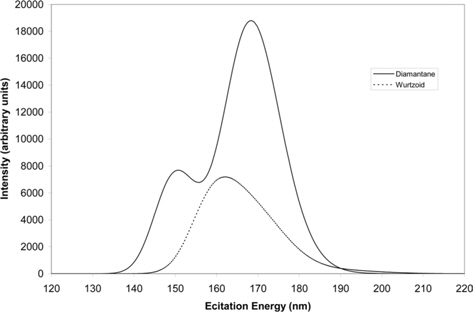
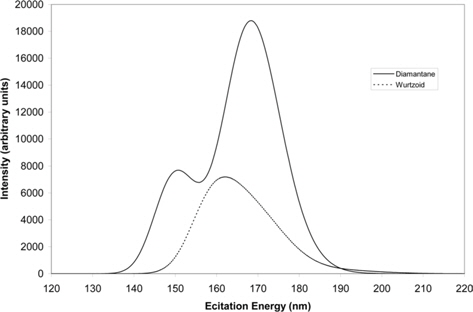
Fig. 9 shows the UV-Vis spectra of diamantane and wurtzoid molecules. These spectra reflect the energy gap and charge transfer trends. The high charge transfer and larger number of hydrogen atoms of diamantane results in higher UV-Vis peaks than those of wurtzoid. The higher energy gap of diamantane (6.26 eV) leads to the first maximum UV-Vis peak position at 151 nm (8.2 eV). The lower energy gap of wurtzoid (5.65 eV) leads to the first maximum UV-Vis peak at 162 nm (7.65 eV). Experimentally, the energy gap can be determined from the UVVis spectra by determining the cut-off excitation energy values, which are approximately 198 nm and 219.4 nm for diamantane and wurtzoid, respectively, as shown in Fig. 9. These correspond to the two gaps of 6.26 eV and 5.65 eV for diamantane and wurtzoid, respectively. Inspection of the two maximums in Fig. 9, that is, 151 nm (8.2 eV) and 162 nm (7.65 eV) for diamantane and wurtzoid, respectively, shows that the two maximums are shifted by approximately 2 eV from their cut-off energies. Although the detailed transition intensities might change this value for other materials, the present shift is approximately constant because it belongs to different phases of the same material (carbon).
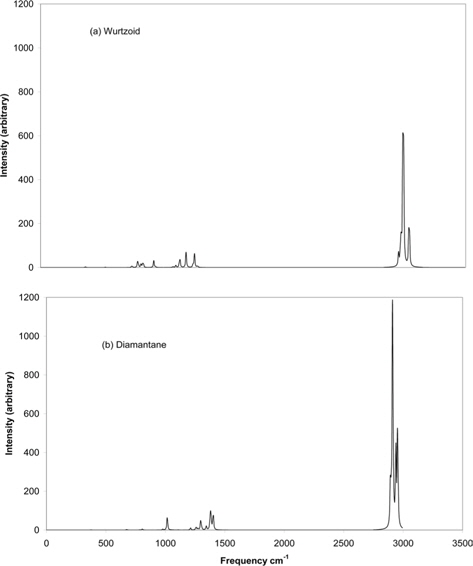
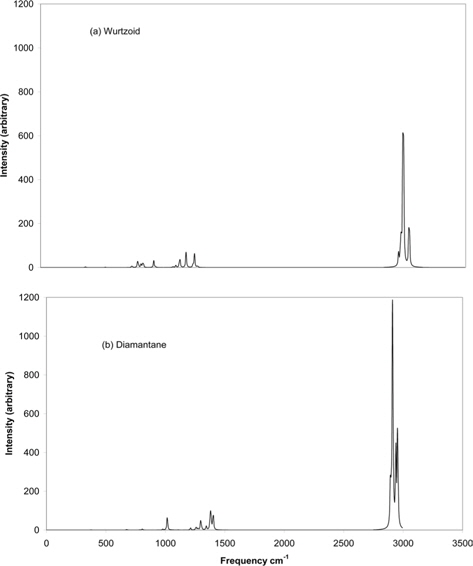
Fig. 10 shows the IR spectra of wurtzoid and diamantane molecules. Since every C atom is passivated by one H atom in wurtzoids, many H vibrations in IR and Raman spectra are lacking or disappear. These vibrations exist in diamondoids. Examples of these lacking vibrations include symmetric and asymmetric stretching, scissoring, rocking, wagging, and twisting modes. These vibrations require the existence of at least two H atoms connected to one C atom. Thus, H vibrations that occur near 3000 cm−1 in wurtzoid have fewer peaks than those of diamantane. The same is true for the range of 1200-1500 cm−1 in which H vibrations are still dominant [16]. The region of 0-1200 is dominated by C vibrations [16]. Since there is a higher number of C-C bonds in wurtzoids, the range of 0-1200 cm−1 in wurtzoids has many peaks that do not exist in diamantane. In addition to the number of peaks, peak movement also occurs. The H vibrations that have frequencies that exceed 3000 cm−1 in wurtzoid become less than 3000 cm−1 in diamantane. This result is due to the effect of the second H atom that exists on some C atoms in diamantane. The highest vibrations of H atoms at wurtzoid surfaces have an approximate reduced mass of 1.09 vs. 1.1 amu for diamantane, which eventually induces higher frequencies according to the following equation:

where
Molecules that can be used as building blocks of hexagonal diamond structures were proposed in this paper. These molecules can be compared with diamondoids, which are the building blocks of the diamond structure. Comparison revealed that hexagonal diamond building blocks have essential differences from cubic diamond blocks, which might explain the differences between the two allotropies at the nanoscale. Our results showed differences in binding energy, bond lengths, tetrahedral angles, band gaps, atomic charges, 1H-NMR spectra, UV-Vis spectra, IR vibrations, etc. Some of the results can be extended to nanocrystals and bulk diamond due to the persistent structural differences between the two phases. The results are in good agreement and consistent with previous experimental results that are mainly available for the diamond structure.

![Binding energy per atom of diamondoids, wurtzoids, and wurtzoid2cs. The results are compared with the experimental binding energy of bulk diamond at 7.55 eV [11].](http://oak.go.kr/repository/journal/15762/HGTSB6_2015_v16n3_192_f003.jpg)
![Energy gap of diamondoids and wurtzoids as a function of the number of carbon atoms. Experimental data for diamondoids, bulk diamond, and hexagonal diamond are from references [1,12,13], respectively.](http://oak.go.kr/repository/journal/15762/HGTSB6_2015_v16n3_192_f004.jpg)