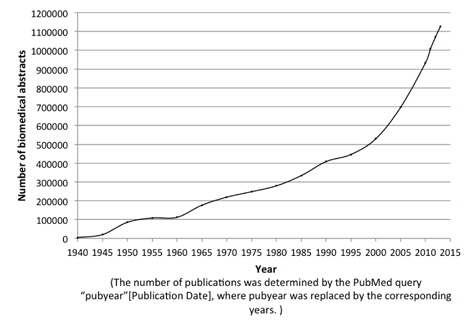
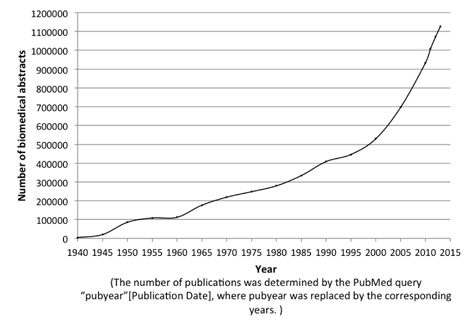
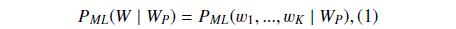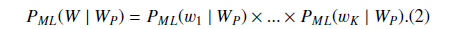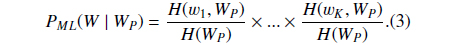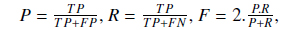The evolution of biomedical text produces a strong demand for automated text mining techniques that can facilitate biomedical researchers to gather and make use of the knowledge in biomedical literature. Figure 1 shows the rapid increase of the number of publications in the MEDLINE [1] database from 1940 until 2014. Protein Named Entity identification is one of the most essential and fundamental predecessor for identification of protein-protein relationships [2, 3], keep protein databases such as UniProtKB [4] up-to-date and many more. However, manual approaches targeting this task are extremely time-consuming, expensive and laborious work, which has led an increasing attention towards automated approaches to help ease these tasks.
Biomedical Named Entity Recognition (BNER) focuses on extraction of words or phrases referring to Biomedical Named Entities (BNEs) in biomedical text and classifying them into appropriate biomedical entity classes such as proteins, genes, DNAs and drug names. However Named Entity Recognition (NER) approaches for biomedical literature do not perform well than those focusing on general text such as newswire domain [5, 6]. BNER is a difficult task because: 1) BNEs contain highly complex vocabulary and are rapidly evolving, 2) most of the BNEs are compound terms and may or may not possess a suffix or a prefix, 3) combination of BNEs may form another BNE, 4) they may have many abbreviations, 5) different aliases can be used to refer the same BNE where the type depends on the context where they exists, etc.
For example “
Much work has been done to develop robust and effective protein identification approaches. They could be classified into dictionary based, heuristic rule based, Machine Learning (ML) based and hybrid approaches. Dictionary based approaches use existing protein dictionaries and/or protein databases to extract and identify proteins in biomedical literature [8]. Due to the different naming schemes referring to the same protein, dictionary based approaches are not effective in identifying them. Even though dictionary based approaches achieve high classification performance, predefined biomedical dictionaries are not able to identify newly introduced protein names.
Rule based approaches use generated rules to identify protein names in biomedical literature [9]. These approaches need domain experts’ knowledge to derive these rules and often time-consuming. ML based BNER approaches mostly use supervised learning algorithms such as Hidden Markov Model (HMM), Support Vector Machines (SVM), Maximum Entropy Model (ME) and Conditional Random Fields (CRFs) [10–16]. However, unsupervised learning models are also proposed [17]. State of the art ML based BNER approaches are dominating the other approaches and can be improved further.
There were several two-step BNER approaches proposed, where extraction of BNEs in text and classification are done in two separate stages. This helps to reduce the training time and also to select more relevant features for each stage [14, 15]. Even though BNEs in text are identified successfully, classifying them into relevant biomedical entity classes still remains as a challenging task. Therefore, identifying protein names is still an open and important task. This paper presents a new method with the highest protein identification performance among BNEs, while significantly reducing the training and testing time.
This paper proposes a statistical feature called Protein-Score, which could be understood as the likelihood that the term “Protein” appears in a MEDLINE abstract with the given BNE. Then, BNEs are denoted by a new set of features including orthographic, keyword, morphological as well as the newly proposed Protein-Score features, and are classified into protein and non-protein entities using Random Forest (RF) [18], a ML technique getting attention lately. A series of experiments is carried out to compare the results with those of the other state of the art approaches. Our Protein Named Entity Recognition (PNER) model based on RF achieved the best performance via experiments on GENIA corpus while significantly reducing the training and testing time.
We discussed the possibility of using the unithood and MEDLINE statistics for protein identification in Sumathipala et al. (2014) [19], where we showed the probabilistic capability of using sub-terms of BNEs to predict whether or not the type of the BNE is a protein. In this study we extend the previous work by further modifying the Protein-Score feature using likelihood measure and introducing new additional features. Here we use the RF machine learning algorithm and show the F-measure value over existing solutions.
The rest of this paper is organized as follows: the next section reviews some related works. In Section 3, we discuss the features used in the study and propose a set of features including the new web-based feature called Protein-Score. In Section 4, we evaluate the effect of the proposed features in identification of Protein Named Entities (PNEs) using RF. We conclude the paper by a summary and directions for future work.
Krauthammer et al. (2000) described a dictionary based system which can automatically identifies gene and protein names in journal articles [8]. Their system was based on BLAST, a popular DNA and protein sequence comparison tool, and on a database of gene and protein names. They achieved a recall of 78.8% and precision of 71.7% for gene and protein name matching.
Seki et al. (2005) proposed a rule based approach for identifying PNEs in biomedical literature with an emphasis on protein boundary expansion [9]. Their method used surface clues to detect potential protein name fragments. They achieved Fmeasure of 63.7% for exact protein matches while achieving F-measure around 81.6% for partial matches. Kuo et al. (2014) proposed a protein name identification model from biological literature and achieved F-measure of 80.6% on GENIA corpus [20]. They used N-gram language model to determine the protein name boundaries and some rules were used to improve the performance. In addition, a dictionary was used to strengthen recognition of abbreviations.
Tater et al. (2009) proposed the use of two different machine learning techniques for protein name extraction [21]. First, they used Bigram language model to extract protein names and then, an automatic rule learning method was used which can identify protein names located in the biological texts. They achieved an F-measure of 66.8% on the GENIA corpus. Zhou et al. (2005) proposed an ensemble of classifiers for protein and gene identification in text, based on a SVM and two discriminative HMMs, which were combined using simple majority voting strategy [10]. They achieved the best F-measure of 82.6% for protein and gene name recognition task.
Finkel et al. (2005) presented a maximum-entropy based approach for identify gene and protein names in biomedical abstracts [11]. They used diverse set of features including “
Yang et al. 2013 presented a two-phase approach for BNER based on semi-Markov Conditional Fields [14]. They used a rich set of features including orthographic, morphological, part-of-speech, features etc. Their experiments based on JNLPBA04 dataset showed 77.7% F-measure for protein name identification. Li et al. 2009 proposed a two-phase BNER approach based on CRFs [15]. First, they identified each BNE with CRFs without considering its biomedical class type and at the second stage they used another CRFs model to determine the relevant class type for each identified BNE. Their experiments achieved overall F-measure of 76.0% for PNE identification on JNLPBA04 corpus. Lin et al. 2004 proposed a hybrid method that uses ME as the ML method incorporated with rule based and dictionary based approaches for post processing [16]. They used orthographic, head noun, morphological and part-of speech features and achieved overall F-measure of 78.5% for protein name identification on GENIA corpus.
Zhange et al. 2013 have proposed an unsupervised learning technique to identify BNEs including proteins [17]. They have achieved F-measure of 67.2% for protein identification on GENIA corpus.
3. Protein Name Identification Features
Feature selection is crucial to the success of ML based PNER systems. In this section, we describe the features used in our system. We utilize orthographic, keyword, morphological features as well as Protein-Score feature based on citations for biomedical abstracts from MEDLINE cited in PubMed. In this paper, we refer both biomedical and non-biomedical named entities as “terms”. A term is composed with one or more “words”such as a single letter, a series of letters, a digit or a series of digits. “word” is called a sub-term if it is not a single letter word or a single digit word. If a term is composed of multiple sub-terms, they are separated by a space or a hyphen.
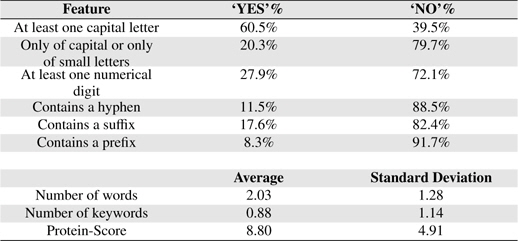
Orthographic features are used to capture knowledge about word orientation such as capitalization, digitalization and other word formation information. These features have been widely used in both biomedical domain [9, 13, 22, 23] and non-biomedical domain [24, 25]. Orthographic features used in this study are: 1) whether or not the term contains at least a capital letter, 2) whether or not the term is composed only of capital letters or only of small letters, 3) whether or not the term contains at least a numerical digit, 4) whether or not the term contains a hyphen and 5) the number of words the term contains.
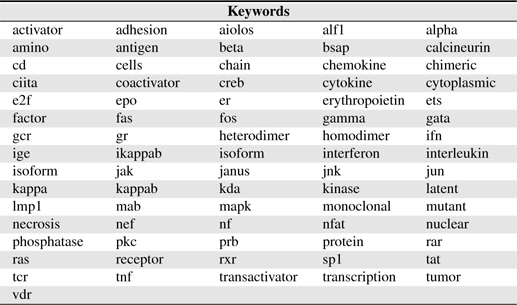
There are many words appearing frequently in BNEs, most of them are compound as mentioned above. Such words (called
[Table 1.] Keywords used in this study

Keywords used in this study
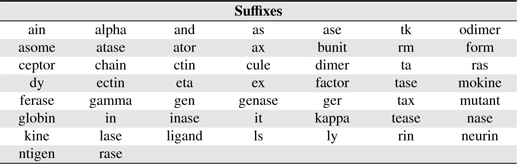
PNEs frequently have a prefix and/or a suffix. Suffixes/prefixes provide important clues for discriminating protein and nonprotein entities. For example BNEs ending with “-
[Table 2.] Prefixes used in this study

Prefixes used in this study
[Table 3.] Suffixes used in this study
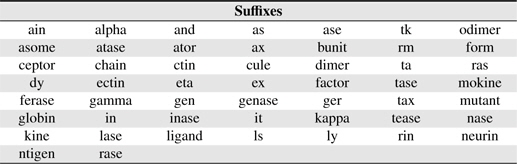
Suffixes used in this study
External features of a term, which are features calculated from data external to the corpus, might provide additional evidential clues for classification of the term. Presence of the term in an external database such as UniProt, external gazetteers and other resources could be an external feature that enhances the performance of BNER. We chose a number of biomedical abstracts from MEDLINE cited in PubMed as the external resource.
Suppose
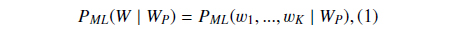
where
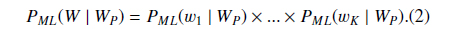
Suppose
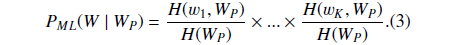
Then we define
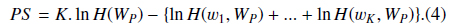
PS could be understood as the likelihood that the abstract with the BNE W is one mention “Protein”. The reason why
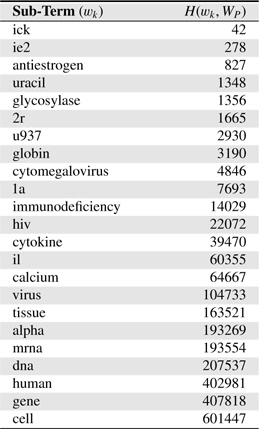
Table 4 shows several sub-terms used in this study and corresponding
[Table 4.] Several sub-terms used and corresponding H(wk,WP) values. H(WP) = 1, 846, 091
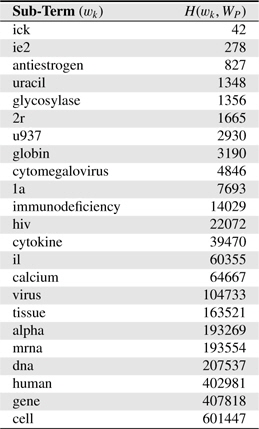
Several sub-terms used and corresponding H(wk,WP) values. H(WP) = 1, 846, 091
[Table 5] Several BNEs and estimated Protein-Score values
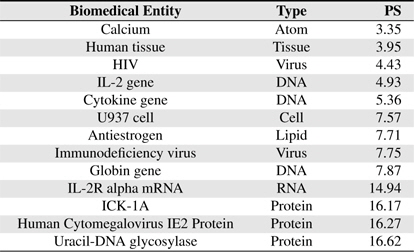
Several BNEs and estimated Protein-Score values
In this section we discussed nine different features including five orthographic, a keyword, a suffix, a prefix and the PS features, which are used to encode BNEs to classify them into protein and non-protein classes using ML algorithms.
4. Implementation and Evaluation
In this section we discuss resources and methods employed in our experiments, following the workflow of ML based PNER model. We also discuss the performance of our PNER system using the proposed features and compared with some of the existing solutions.
In our experiments, Weka 3.6.12 was used to implement the ML algorithm [26]. GENIA corpus was used as the lexical resource of BNEs during training and testing. It contains MEDLINE abstracts, selected using a PubMed query for the three MeSH (Medical Subject Headings) terms “
PubMed was used to access the MEDLINE database for biomedical abstracts. It is a free resource which comprises more than 24 million citations for biomedical literature from MED-LINE, life science journals and online books [27]. MEDLINE covers journal citations and abstracts for biomedical literature from around the world [1].
Hardware configuration we used are as follows: Intel ®Core TM i7 CPU, clock speed 3.50 GHz, Memory (RAM) 32 GB 1600 MHz DDR3, System type: OSX Version 10.9.5 Operating System.
We carried out experiments to classify BNEs annotated in GENIA corpus into protein and non-protein classes. We have extracted 92,512 BNEs in total from GENIA including 34,221 PNEs.
Table 6 shows some statistical information of the nine features of BNEs extracted from GENIA.
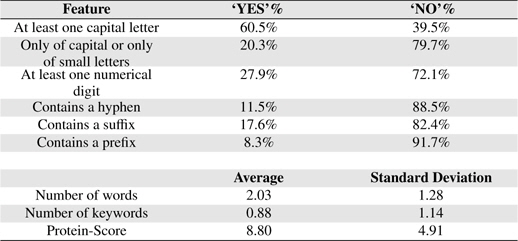
Feature Statistics
BNEs in GENIA corpus with protein annotations, which are enclosed by XML semantic tags prefixed with G#protein are considered as PNEs and all others as non-protein named entities. These non-protein named entities belong to different biomedical entity classes such as cell, atom, DNA, Virus, RNA. Ten-fold cross validation was used to evaluate the ML algorithm. In each iteration, nine-tenth of BNEs was used for training and the rest was used for testing.
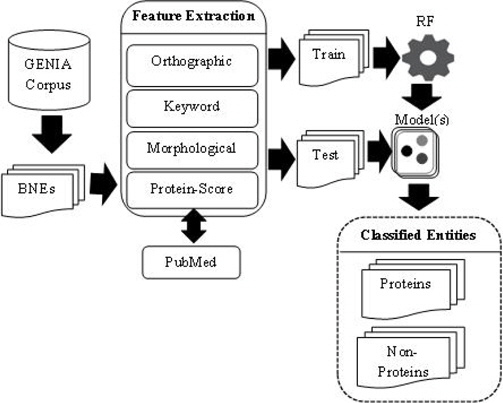
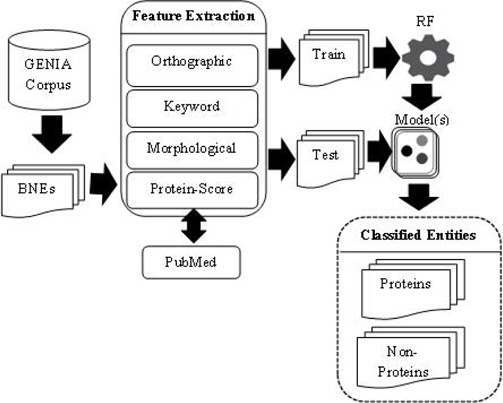
PS feature plays a vital role in our PNE identification model. In training and testing PS was estimated using more than 24 million biomedical abstracts in MEDLINE accessed through PubMed. Total number of sub-terms taken from all the BNEs are 8, 247. Average number of sub-terms in a PNE is around 2. We employed well-known RF machine learning technique with the nine proposed features. In Table 7, we summarize the results from RF algorithm. Rows of the confusion matrix in Table 7, correspond to actual classes and its columns correspond to the predicted classes. Figure 3 shows the overall architecture of our proposed approach.
[Table 7.] Testing results on the GENIA corpus by our approaches. a: Protein, b: Non-Protein

Testing results on the GENIA corpus by our approaches. a: Protein, b: Non-Protein
Ensemble methods have become a popular and widely used tool within the past few years in the field of bioinformatics, because they are applicable in high dimensional problems with complex interactions [28–31]. In the study of [19], we conducted experiments using several ML techniques, and RF performed the best. RF is a powerful classification algorithm in the group of ensemble learning and obtains growing attention on these days. It uses an ensemble of unpruned Decision Trees called CART [32], each of which is constructed on bootstrap sampling of the training data set based on randomly selected subset of features [18]. The major parameters of RF are the number of trees (
In order to analyze the impact of various other techniques and compare the ultimate results of our approach with other existing solutions, we use common evaluation measures: precision (P), recall (R) and F-measure (F). These measures are formulated as follows:
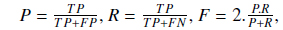
where TP is the number of true positives retrieved, FP is the number of false positives retrieved and FN is the number of false negatives.
In our experiments, RF classifier achieved the best results yielding the overall precision, recall and F-measure values of 92.7%, 91.7% and 92.2% respectively, with the overall classification accuracy of 94.3%. RF has taken 65.6 seconds for training of an iteration of the ten-fold cross-validation.
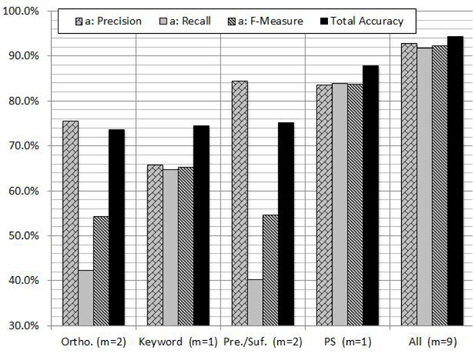
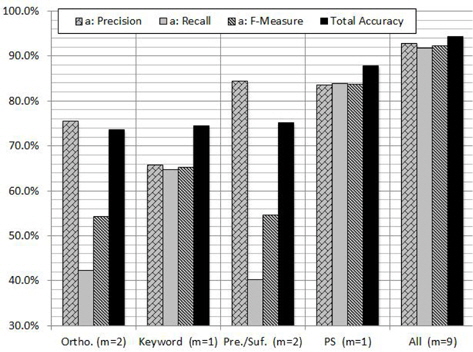
In order to evaluate the contribution of each feature and subset of features, we carried out experiments on GENIA corpus using RF algorithm. Figure 4 shows the contribution of each feature to the PNER task. According to the figure we can notice that the PS feature plays a vital role in increasing all the measures. As shown in Figure 4, the highest classification performance was obtained with the proposed combination of features: orthographic, keyword, morphological and PS features. When we use PS feature individually, the
PS can be considered as a dynamic feature because it is based on the number of MEDLINE abstracts cited in the PubMed database, which are gradually increasing. In Sumathipala et al. (2014) [19] we introduced a unithood measure called Proteinhood which quantified the dependency between sub-terms of biomedical term candidates by measuring the probabilistic strength of forming a PNE. Proteinhood values were estimated using the protein sub-terms in the training data set. Therefore the measure might not be effective in identifying PNEs if their sub-terms are not in the training data.
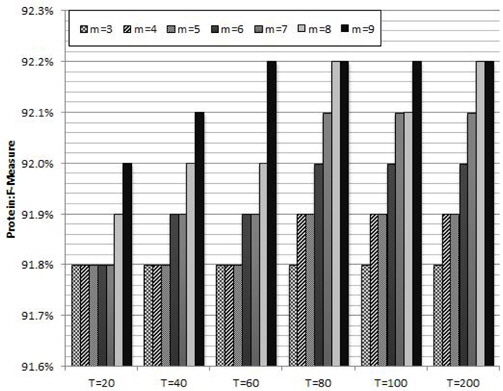
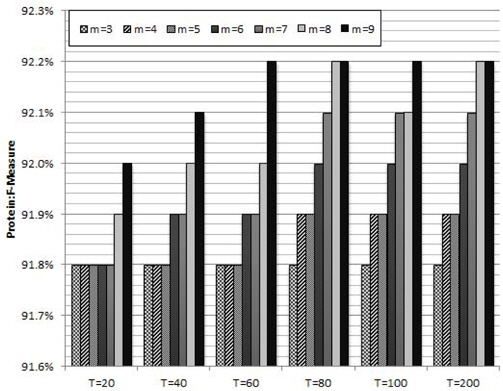
Figure 2 shows the average protein F-measure values of ten trials with different combinations of the parameters in RF:
The reason of the inconsistency would be that the assumption does not hold in the problem: the contribution of the features is distributed almost equally among them. Instead, as seen in Figure 4, the only feature PS has a large contribution to the classification in the case [18].
In general, direct comparison of protein name identification methods is challenging because some classify several kinds of BNEs and the others do not as well as there is a wide variation of both entity classes and test sets used by each research group. In many studies, PNE classification was conducted as a part of large systems aiming to extract BNEs from biomedical literature and to classify them into several biological classes.
However, the highest performance of our approach shown in Table 8, would never be underestimated, because protein identification is an important task in the molecular biology domain and the approach is itself could be incorporated in a large scale BNER system as well as the same idea of PS could be introduced into BNEs other than proteins.
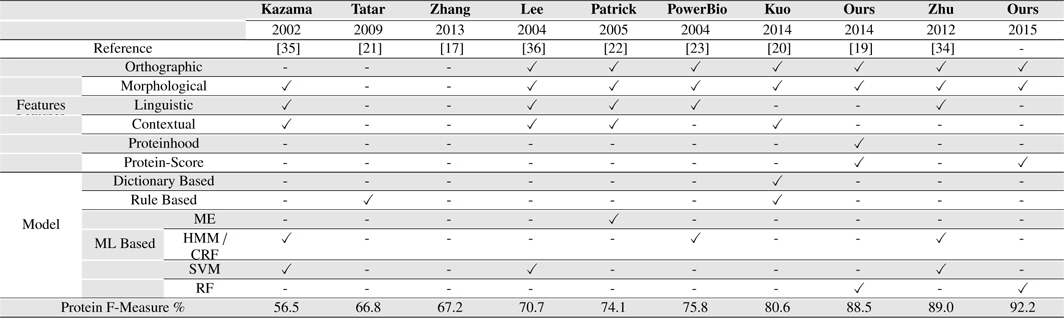
[Table 8.] Our Systems Compared with several existing BNER approaches on the GENIA corpus
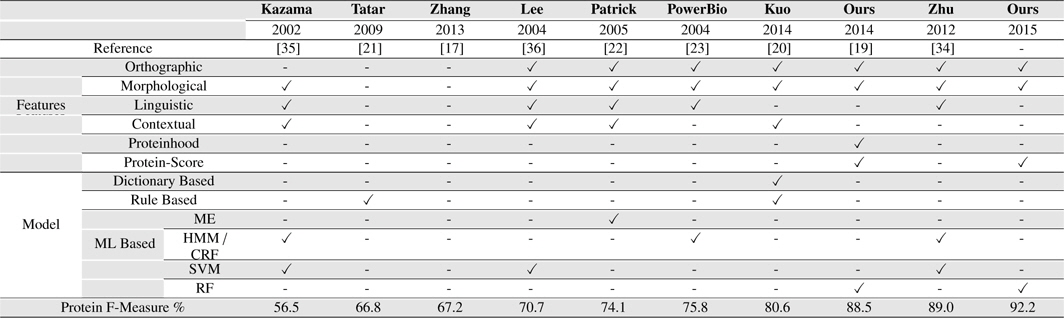
Our Systems Compared with several existing BNER approaches on the GENIA corpus
Our approach outperformed all the other solutions on GENIA corpus, achieving an F-measure of 92.2% for PNER task. It presents an improvement of 3.12% over the second best system we compared, Zhu et al. (2012) which achieved the F-measure of 89.0% on GENIA corpus for PNE identification task [34].
In our approach, we used RF with a small number of features to identify proteins. The experiments show that, our approach takes short training time which shows that it is efficient, effective and economically beneficial.
In this paper, we presents a PNER approach based on a new set of features including orthographic, morphological and PS features. Our approach outperforms the other state of the art BNER methods, in the view point of protein name identifi- cation. We achieved the best performance, which proves the importance of features we used in protein name identification task. We demonstrated the effectiveness of our approach on GENIA corpus, and make comparisons with some related tasks.
Our future work is focused on extending the classification into more biomedical classes including “DNA”, “RNA”, “CELL-LINE” and “Cell-TYPE”.
Conflict of Interest
No potential conflict of interest relevant to this article was reported.