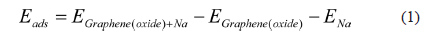Lithium ion batteries (LIBs) are essential energy storage systems present in a number of devices used in our daily lives such as laptops and mobile phones. However, the developments made in energy storage technology have outpaced the increase in energy storage needs [1]. Energy storage technologies such as Li batteries are considered as alternatives to fossil fuels, which are rapidly depleting all over the world. However, because Li is a scarce resource on Earth, producing large-sized LIBs is limited by the exponential increase in its cost [2]. In this context, sodium ion batteries (SIBs) are excellent alternatives to LIBs because of their lower cost and the greater availability of Na compared to Li. In fact, Na is abundantly available and the de-insertion behavior of Na in SIBs is very similar to that of Li in LIBs [3]. Hard carbon materials that have graphite layers and amorphous regions are widely used as anodes in SIBs [4]. In addition, carbon black, amorphous carbon, mesoporous carbon, and carbon fiber are being experimentally tested as anode materials for SIBs [5]. Among the various carbon materials, graphene, which has excellent electrical characteristics and physical properties, is gaining attention as an anode material candidate for SIBs. In a recent study, David et al. [6] synthesized layered freestanding papers comprised of acid-exfoliated few-layer molybdenum disulfide (MoS2) and reduced graphene oxide (rGO) flakes for use as self-standing flexible electrodes in SIBs. The electrode showed a good Na cycling ability with a stable charge capacity of approximately 230 mAh g-1 taking into account the total weight of the electrode and exhibited coulombic efficiencies up to approximately 99%. In addition, static uniaxial tensile tests performed on the crumpled composite papers showed high average strains to failure up to approximately 2%. Xie et al. [7] synthesized SnS2 nanoplatelets at graphene nanocomposites using a morphology controlled hydrothermal method. When SnS2/graphene nanosheets were used as anode materials in SIBs, a high reversible specific Na-ion storage capacity of 725 mAh g-1, stable cyclability, and an enhanced high-rate capability were observed.
Although such studies exist, the detailed mechanisms on Na adsorption on graphene and graphene oxide have not been fully elucidated yet. Therefore, in this work, we investigated the Na adsorption mechanisms on graphene-based anodes used in SIBs at the atomistic level by analyzing the adsorption energies and electronic properties using density functional theory (DFT).
The DFT calculations were performed using the Vienna Ab-initio Simulation Package (VASP) [8,9] with the projector augmented wave (PAW) method [10], and generalized gradient approximation (GGA) and Perdew-Burke-Ernzerhof (PBE) functionals [11,12]. The PBE functional has been used to successfully describe the interactions between an adsorbate and various surfaces [13-15] including sp2 carbon-based materials [16-19].
The energy cut-off was set at 500 eV, and the cell size was 9.840 × 9.840 × 15 Å. The calculations were performed under the periodic boundary conditions, with the vacuum separation distance set to 15 Å along the z-direction, to avoid interactions between the images. In the energy and electronic property calculations, k-point samplings for the Brillouin zone were performed using the 5 × 5 × 1 Monkhorst-Pack k-point mesh [20]. The atomic positions were optimized until the change in energy was less than 1 × 10-6 eV per cell, and the force on each atom was less than 0.01 eV Å-1. Spin-polarization and dipole corrections were also incorporated into the calculations. The adsorption energies of Na-adsorbed graphene and graphene oxide were defined by Eq. (1):
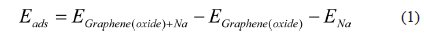
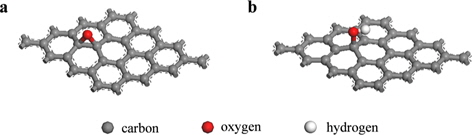
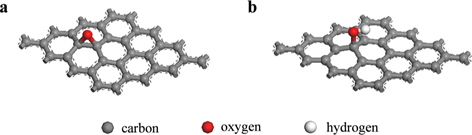
, where EGraphene(oxide)+Na is the total energy of the Na-adsorbed graphene or graphene oxide; EGraphene(oxide) is the total energy of the graphene or graphene oxide, and ENa is the total energy of the isolated Na. Dispersion corrections [21] were also considered in all the calculations. Charge transfer was calculated using the density derived electrostatic and chemical (DDEC) method [22,23]. Graphene oxide was assumed to consist of epoxide and hydroxyl oxidation functional groups, and each group was introduced to the pristine graphene shown in Fig. 1.
3.1. Adsorption energies and charge analysis
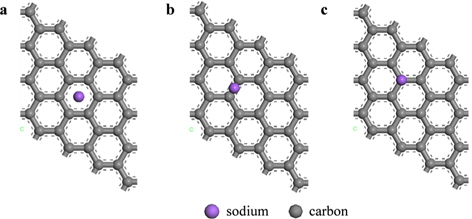
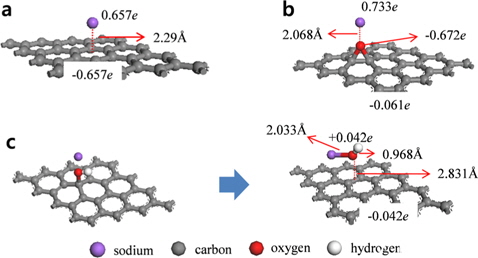
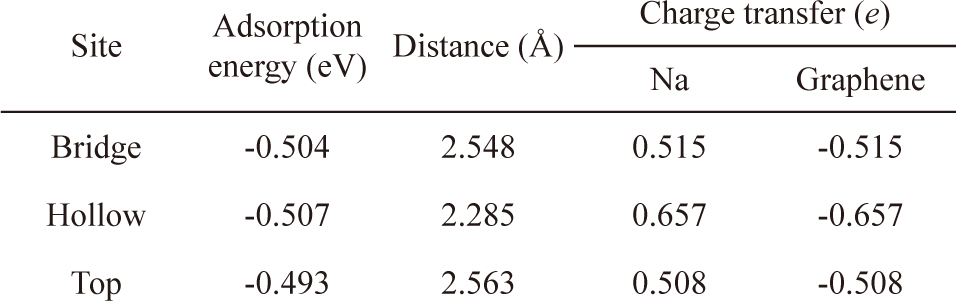
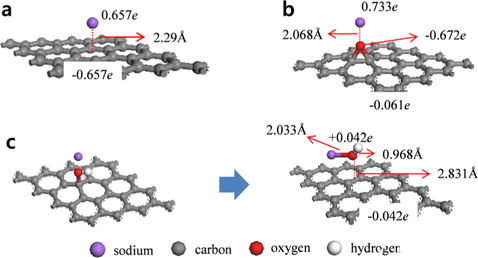
Na atoms were adsorbed onto hollow, bridge, and top sites of pristine graphene to determine the most stable position (Fig. 2). The hollow site was the most stable position for Na atoms, and the adsorption energy was -0.507 eV. After Na adsorption, the optimized distance between the graphene and Na was 2.29 Å. The bridge sites were the second most stable adsorption sites for Na with an adsorption energy of -0.504 eV, whereas adsorption onto the top sites was the least stable with an adsorption energy of -0.493 eV. These results were confirmed by charge analysis showing that at all three sites, the charge was transferred from the Na to the graphene after adsorption, and the number of DDEC charges of the graphene at the hollow, bridge, and top sites was -0.657
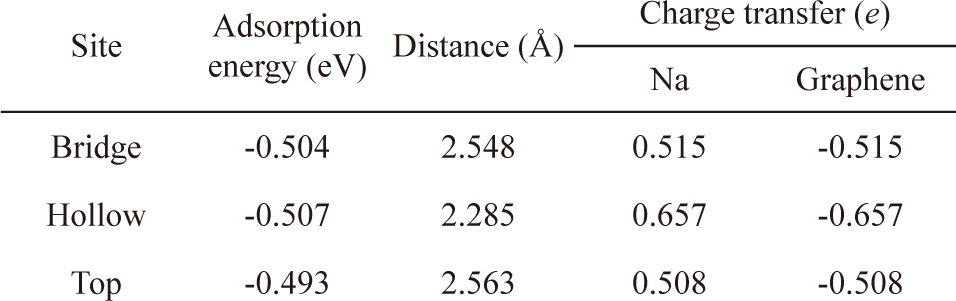
Adsorption energy, distance between Na atoms and graphene, and charge distribution of the bridge, hollow, and top sites on the graphene
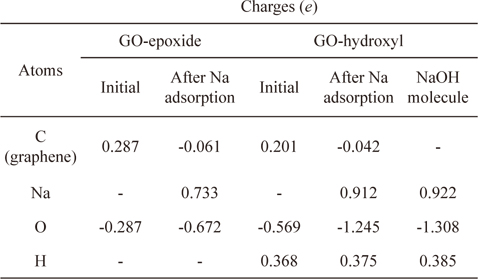
Graphene oxide was assumed to contain epoxide and hydroxyl functional groups, and Na was adsorbed onto each of these types of functional groups on the graphene oxide. First, Na was adsorbed vertically onto the oxygen atoms of the graphene oxide-epoxide (GO-epoxide). The adsorption energy for this case was -1.024 eV, which was approximately twice the value for the adsorption of Na onto the pristine graphene. We believe that the oxygen in the epoxide caused a doping effect, and subsequently, the adsorption energy was enhanced. In addition, the optimized distance between the graphene and Na was 2.068 Å. The optimized structure of the Na-adsorbed GO-epoxide and the charge transfer values for the graphene oxide and Na-adsorbed graphene oxide are summarized in Fig. 3b and Table 2. The sum of the charges of the oxygen and carbon in the GO-epoxide was -0.287
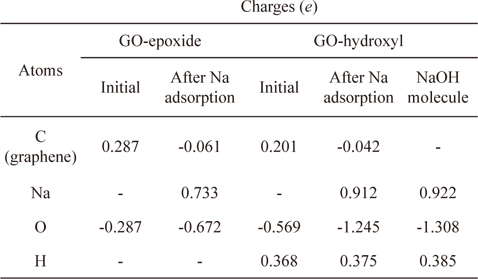
Charge distribution on the Na-adsorbed graphene oxide (epoxide and hydroxyl) compared with those on the pristine graphene oxide (epoxide and hydroxyl) and NaOH
Next, Na was adsorbed vertically onto the oxygen atom of the hydroxyl group in the graphene oxide-hydroxyl (GO-hydroxyl). Unlike graphene and GO-epoxide, the hydroxyl group dissociated as a result of Na adsorption and produced NaOH as a by-product (Fig. 3c). The optimized distance between the oxygen of the NaOH and graphene was 2.831 Å, and the adsorption energy between the NaOH and graphene was -0.402 eV. Kim et al. [24] showed that the epoxide groups are much more stable than the hydroxyl groups in graphene oxide by DFT with the GGA-PW91 functional. Because of the low binding energy, a hydroxyl group in the interior aromatic domain of the graphene oxide is unstable, and as a consequence, the hydroxyl group can be dissociated [25,26]. However, it should be noted that because the present investigation is based on DFT calculations that focus on the ground state of Na-graphene systems, the results obtained during the operation of SIBs may be different. Dissociated NaOH has a similar charge distribution as an isolated NaOH molecule. As a result, charges of -0.042
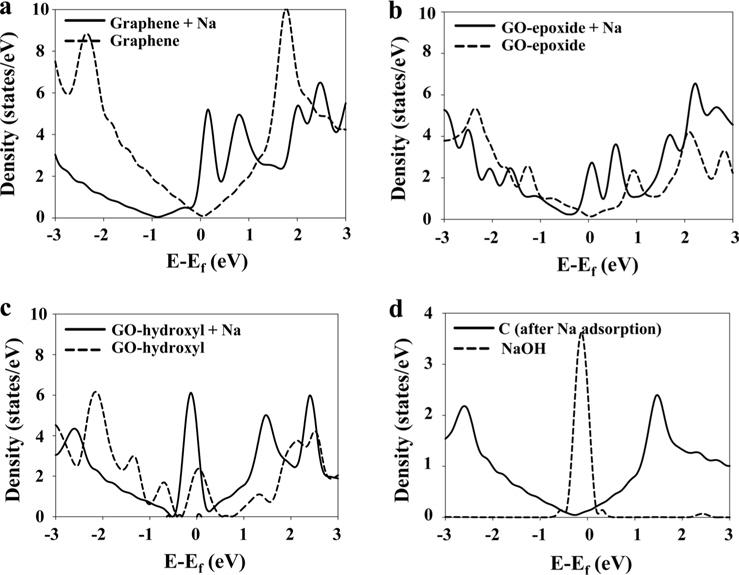
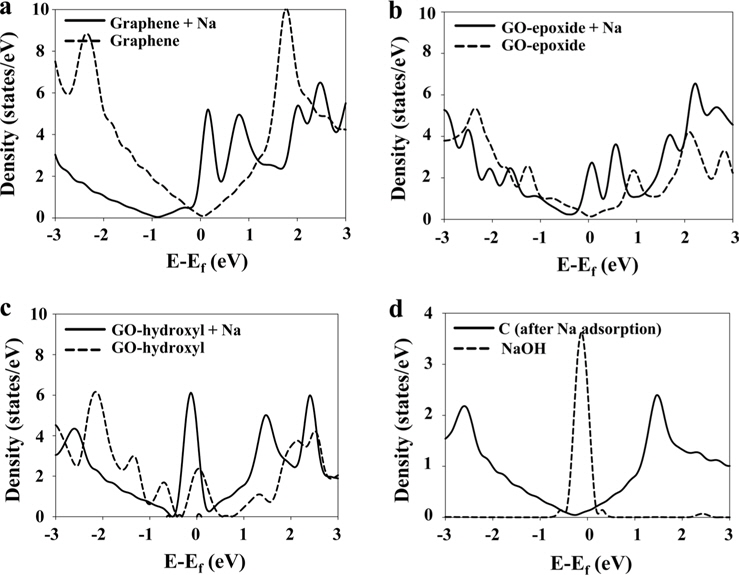
We investigated the electronic properties of the graphene and graphene oxide by analyzing the density of states (DOS). The Fermi levels for all the DOS shifted to zero. The DOS of pristine and Na-adsorbed graphene are shown in Fig. 4a. After Na adsorption, however, the DOS of the Na-adsorbed graphene shifted down, and their intensity near the Fermi level increased, indicating that more electronic states can exist at the Fermi level. This could be attributed to the fact that electrons from Na are injected towards the graphene.
Fig. 4b shows the DOS of the GO-epoxide and Na-adsorbed GO-epoxide. The Dirac point was not observed in the DOS of the GO-epoxide because the 2
Fig. 4c shows the DOS of the pristine GO-hydroxyl and Na-adsorbed GO-hydroxyl. While the DOS of the GO-hydroxyl exhibited several peaks between -2 and 2 eV, the DOS of the Na-adsorbed GO-hydroxyl exhibited a broad line between -2 and 2 eV, similar to the case of the graphene. In addition, the intensity of the DOS near the Fermi level was significantly increased after Na adsorption. To investigate this in more detail, we calculated the local density of states (LDOS) of the carbon (graphene) after Na adsorption and the NaOH molecules (Fig. 4d). The LDOS of the carbon, shown in Fig. 4d, was similar to that of the graphene. Therefore, we concluded that the GO-hydroxyl reverted to the graphene, as a result of the dissociation of OH. In addition, a broad line between -2 and 2 eV in the DOS of the Na-adsorbed GO-hydroxyl was a result of the LDOS of the carbon. In the case of the LDOS of the NaOH molecules, the DOS of the NaOH exhibited a huge peak at the Fermi level, providing the energy levels of the molecular orbitals for the NaOH molecules rather than the band structure. Therefore, in Fig. 4c, the intensity of the DOS peak around the Fermi level was significantly increased after Na adsorption, which appears to be affected by the DOS of the dissociated NaOH.
As a result, the DOS of the Na-adsorbed graphene shifted down more than that of the GO-epoxide, and the intensity of the DOS around the Fermi level was higher than that of the Na-adsorbed GO-epoxide. Although the adsorption energy for Na onto GO-epoxide was higher than the corresponding value for graphene, after Na adsorption, the electronic properties of the graphene were better than those of the GO-epoxide because oxygen doping weakens the sp2- hybridization of the carbon atoms in the GO. In the case of the GO-hydroxyl, after Na adsorption, the OH group dissociated and produced NaOH molecules. In addition, the dissociated NaOH molecules were adsorbed onto the graphene by weak physical adsorption.
In this paper, we investigated the adsorption energies and electronic properties for the adsorption of Na onto graphene and graphene oxide, using the first-principles DFT method. Na adsorption was the most stable at the hollow sites of pristine graphene with an adsorption energy of -0.507 eV. In the case of the GO-epoxide, the Na adsorption energy was -1.024 eV, which was more negative than the adsorption energy for Na onto pristine graphene. In the case of the GO-hydroxyl, the hydroxyl group dissociated from the GO-hydroxyl, and NaOH was formed as a by-product. After Na adsorption, the DOS of both graphene and GO-epoxide shifted down, and the intensity of the DOS around the Fermi level increased due to electron injection in both cases. In addition, it was found that the DOS of the Na-adsorbed graphene shifted down more than that of the GO-epoxide, and the intensity of the DOS around the Fermi level for the Na-adsorbed graphene was higher than that for the Na-adsorbed GO-epoxide. Therefore, after Na adsorption, the electronic properties of the graphene were better than those of the GO-epoxide because of oxygen doping which weakened the sp2-hybridization of the carbon atoms in the GO.