Non-small-cell lung cancer (NSCLC) is one of the leading causes of cancer death worldwide [1]. Various molecules, including flavonoids, have been reported to be useful in treating NSCLC [2-3, 4]. Quercetin, a polyphenolic flavonoid, is found in many medicinal plants such as Euonymus alatus, Thuja occidentalis, etc. and in edible foods such as red onions, apples, broccoli, etc [5]. Quercetin has already been reported to be beneficial in preventing several cancers, including lung cancer [4, 6]. However, the mechanism of action of this chemopreventive flavonoid, quercetin, has never been studied in details, especially against NSCLC.
Increased synthesis of inflammatory cytokines in the tumor’s microenvironment helps NSCLC cells to maintain their survivability against all types of diverse conditions [7, 8]. Interleukine-6 (IL-6) is one of the major cytokines that promotes inflammatory responses in the tumor’s microenvironment [9]. IL-6 helps cancer cells to proliferate in an uncontrolled manner by ignoring apoptotic signals [7, 8]. Activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a key upstream effector molecule of IL-6 [10], is directly linked with elevated titer of IL-6. Therefore, this particular cytokine, IL-6, and its upstream effector molecule, NF-κB, are found to be over expressed during uncontrolled cell proliferation and cell survivability of NSCLC cells.
IL-6 activates several downstream signaling molecules, including signal transducer and activator of transcription 3 (STAT3), that in turn promote inflammatory responses and help the cancer cells to maintain proper homeostasis [11], thus accelerating the lung tumorigenesis process [12, 13]. On the other hand, up-regulation of major signaling molecules such as NF-κB and STAT3 is known to maintain the regulation of the B-cell lymphoma 2 (Bcl2) family proteins or more specifically the expression of a major anti-apoptotic gene, Bcl2 [14, 15]. An up-regulated Bcl2 synthesis, thus, helps in maintaining its ratio to the Bcl-2 Antagonist X (Bax) molecule, which, therefore, retains the mitochondrial integrity that is needed for the survivability of cancer cells [16, 17].
Flavonoids, especially quercetin, have already been reported to be major molecules that reduce inflammation [18, 19]. Therefore, in this study, our main objective was to ascertain whether quercetin had any profound role in reducing the major inflammatory cytokine, IL-6, and its associated signaling molecules in a NSCLC cell line, A549. Because this NSCLC cell line, A549, was found to bear drug resistance due to a mutation in the kras gene, the effectiveness of quercetin, if any, on this particular cell line would be of great significance. Apart from this, whether or not quercetin induced modulations of IL-6 and its associated signaling molecules were able to bring about apoptotic death of A549 cells was also studied in this investigation.
Dulbecco’s modified eagle medium (DMEM), roswell park memorial institute (RPMI)-1640 medium, fetal bovine serum (FBS), antibiotics, trypsin and ethylenediaminetetraacetic acid (EDTA) were purchased from Gibco BRL (Grand Island, NY, USA). Tissue culture plastic wares were obtained from Tarsons, India. Quercetin was obtained from Himedia, India.
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), propidium iodide, 4’, 6-diamidino-2-phenylindole (DAPI), rhodamine 123 and all secondary antibodies were purchased from Sigma Aldrich, USA. Annexin V-fluorescein isothiocyanate (FITC), anti-NF-κB, anti-STAT3, anti-Bax, anti-Bcl2, anti-caspase 3 and anti- Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) monoclonal antibodies were purchased from Santa Cruz Biotechnology Inc., USA. The primary antibodies of IL-6, cytochrome c, and FITC conjugated secondary antibodies were obtained from BD Bioscience, USA. Anti-Bromodeoxyuridine (BrdU), anti-p-STAT3 and anti-poly ADP-ribose polymerase (PARP), anti-bodies was procured from Cell Signaling, USA.
Human NSCLC cell lines, A549 and H460, were collected from the National Centre for Cell Science, Pune, India. Cells were cultured in Dulbecco’s modified eagle medium (DMEM, A549) and Rosewell park memorial institue (RPMI-1640, h460) media with 10% heat inactivated FBS and 1% antibiotic (PSN) and were maintained at 37ºC with 5% CO2 in a humidified incubator.
Cells were harvested with 0.025% trypsin and 0.52-mM EDTA in phosphate buffer saline (PBS).
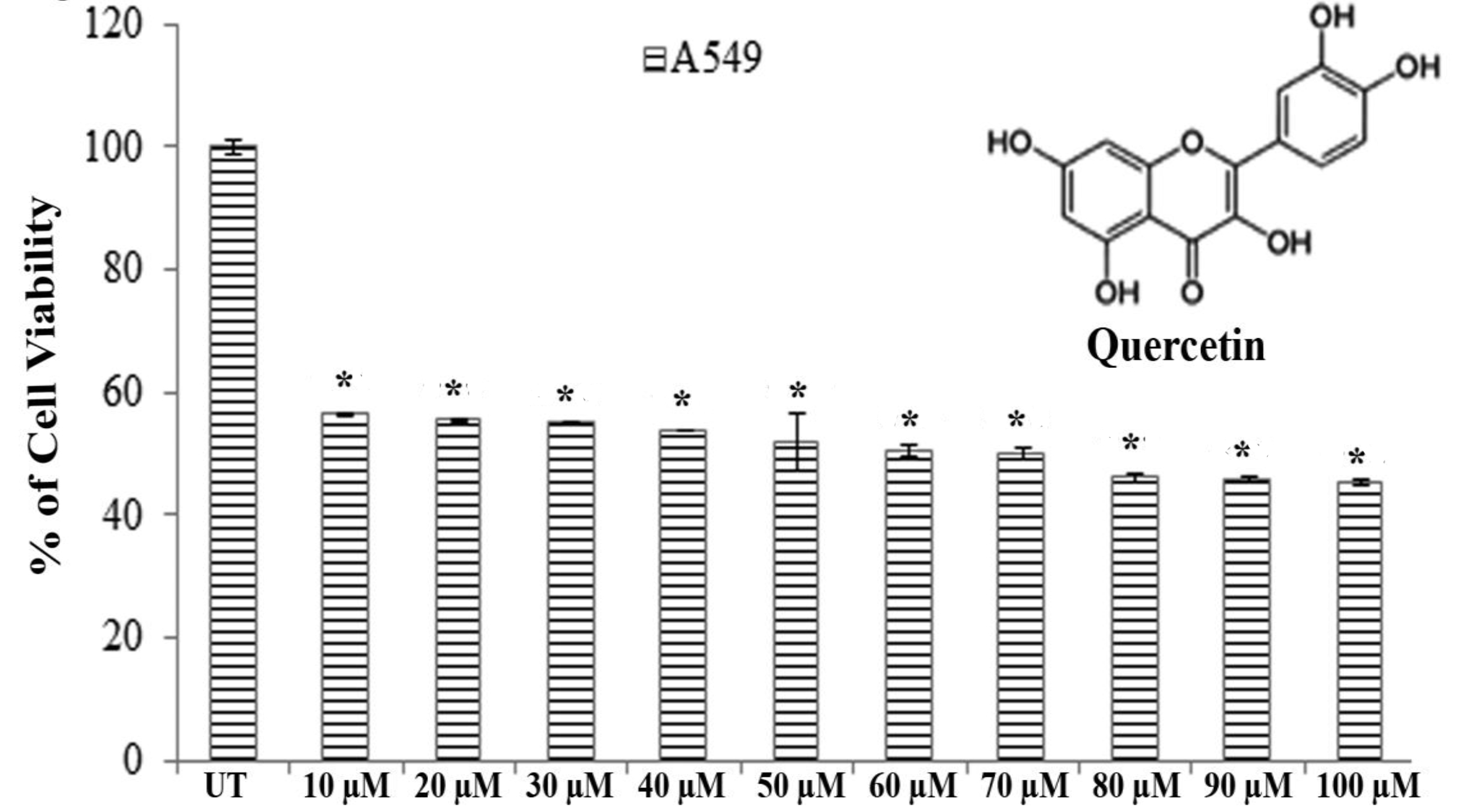
A549 and H460 cells (1 × 102 cells per well) were seeded in 96 well plates. They were allowed to settle for 24 hours before treatment. Then, the cells were treated with quercetin (10 – 100 μM) for 24 hours with 5% CO2 at 37ºC. Cell viability was determined by using the MTT assay [20]. The concentration that reduced the cell viability by 50% was taken as the IC50 dose.
Both quercetin treated and untreated A549 cells were collected and fixed, rinsed twice with PBS, and stained with 10-μM DAPI for 30 minutes to determine DNA nick generation, if any, under a fluorescence microscope (Axiovert CFL 40, Zeiss). DNA strand breaks were analyzed by using the TUNEL assay with a flow cytometer (FACS Calibur, BD Bioscience), following the method of Darzynkiewicz
After treatment with 66 μM of quercetin for 12, 18, 24, 36 and 48 hours, cells were harvested with trypsinization and were labeled with Annexin V (100 ng/mL) and propidium iodide (PI, 50 μg/mL) at room temperature.
Stained cells were incubated for 15 minutes in the dark and were analyzed using a flow cytometer (FACS Calibur, BD Bioscience, USA), taking a minimum of 10,000 cells in each sample.
Cells, 2 × 104, were seeded and treated with or without quercetin (66 μM) for different time points (12, 18, 24, 36 and 48 hours).
Cell cycle analysis was done using a fluorescence activated cell sorter (FACS) calibur flow cytometer (BD Bioscience, USA) by following the standard protocol [5].
Changes in the mitochondrial membrane potential after quercetin treatment were examined at the 6-, 12-, 18- and 24-hours time points after the live cells had been stained with 20-μM rhodamine 123.
The mean fluorescence intensity was detected at excitation and emission wavelengths of 511 nm and 534 nm, respectively, by fluorimetry (Perkin Elmer, USA).
A549 cells, 2 × 103, were treated with drugs at different time points between 15 and 60 minutes and between 2 and 6 hours to check the activity of NF-κB. The control cells received no drugs. The cells were pelleted down, washed with PBS, and the pellet was re-suspended in l mL of 1X HEPES Triton-X 100 buffer (10-mM HEPES, 5-mM MgCl2, 320-mM sucrose, pH 7.4, 1% Triton X-100). The cells were then incubated at 4ºC for 10 minutes, pelleted down, and resuspended in 1% PBS-bovine serum albumin (BSA) solution. Thereafter, cells were incubated with NF-κB antibody (1 : 100) for 30 minutes at 4ºC, washed with 1% PBSBSA solution and then pelleted down. Three hundred μL of PBS-BSA and 5 μL of PI (50 μg/mL) were added to the pellet for nucleus staining, and the pellets were kept in ice for 10 minutes.
Finally, the cells were analyzed by using FACS Calibur with ‘CELL Quest’ software (Becton Dickinson, San Jose, CA, USA).
Quercetin treated (12, 18 and 24 hours) and untreated cells were stained at 4°C in the dark for 20 minutes with FITC conjugated anti-human IL-6 antibody. Intracellular IL-6 titer was detected in the cells by fixing and permeabilizing with a Cytofix/Cytoperm Kit (BD Pharmingen, San Diego, CA, USA). Briefly, cells were incubated in culture with Monensin (BD Pharmingen, San Diego, CA, USA) for 8 hours prior to harvesting to prevent the transport of proteins mediated through Golgi bodies in order to capture them inside the cells. For intracellular staining, cells were harvested and pelleted down. The cell pellet was washed with PBS, and the centrifugation step was repeated. After that, cells were incubated with Cytofixation/Cytopermeabilization (BD Pharmingen, San Diego, CA, USA) solution at 4°C in the dark for 20 minutes. Then, the cells were washed, and intracellular staining was performed in a washing buffer containing saponin. Intracellular proteins were stained with purified FITC conjugated anti-human IL-6 antibody.
The cells were fixed in 1% paraformaldehyde and analyzed by using a FACS Calibur with ‘CELL Quest’ software (Becton Dickinson, San Jose, CA, USA).
For the p-STAT3 expression assay, A549 cells (2 × 103 cells/mL) were treated with IL-6 (10 ng) for 15 minutes after quercetin induction, or they were treated only with quercetin. The controls received no drug. Then, the cells were trypsinized and pelleted down. Both untreated and treated cell pellets were incubated with 1% BSA solution for 1 hour. Cells were then washed with 0.05% Triton-XPBS solution and incubated with primary antibody (1 : 200) of specific protein and then labeled with FITC tagged secondary antibody (1 : 100).
Thereafter, cells were analyzed by using flow cytometry to obtain specific protein activity percentages. Ten thousand events were analyzed for each sample using an appropriate gating to select single cell population; the same gate was used for all the samples, and the p-STAT3 positive cell population was analyzed by using a FACS Calibur (BD Bioscience, USA) with ‘CELL Quest’ software (Becton Dickinson, San Jose, CA, USA).
Cells treated with quercetin and cell not treated with quercetin were collected, washed twice with ice cold PBS. Then, the total and the cytosolic proteins were collected and washed twice with ice cold PBS.
Then, the total and the cytosolic proteins were isolated from the extract and stored at 20°C according to the method of Ganguly
For western blot analyses, equal amounts of protein were loaded, and samples were denatured in 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS– PAGE) for STAT3, p-STAT3, and PARP and in 15% SDSPAGE for Bax, Bcl2, cytochrome c and caspase 3, following the method of Sambrook and Russell [23]. GAPDH was used as a housekeeping gene control.
Quantification of proteins was done densitometrically using ‘image J’ software. Western blots of proteins of interest were analyzed in three separate sets of experiments.
Unless otherwise stated, all the data reported were arithmetic means of data from independent experiments performed in triplicate. Results were expressed as means ± SDs (standard deviations). Statistical analyses were performed by using the one-way analysis of variance (ANOVA) with the least significant difference (LSD) post-hoc test by using the SPSS 16 software. Statistical significance was considered as *
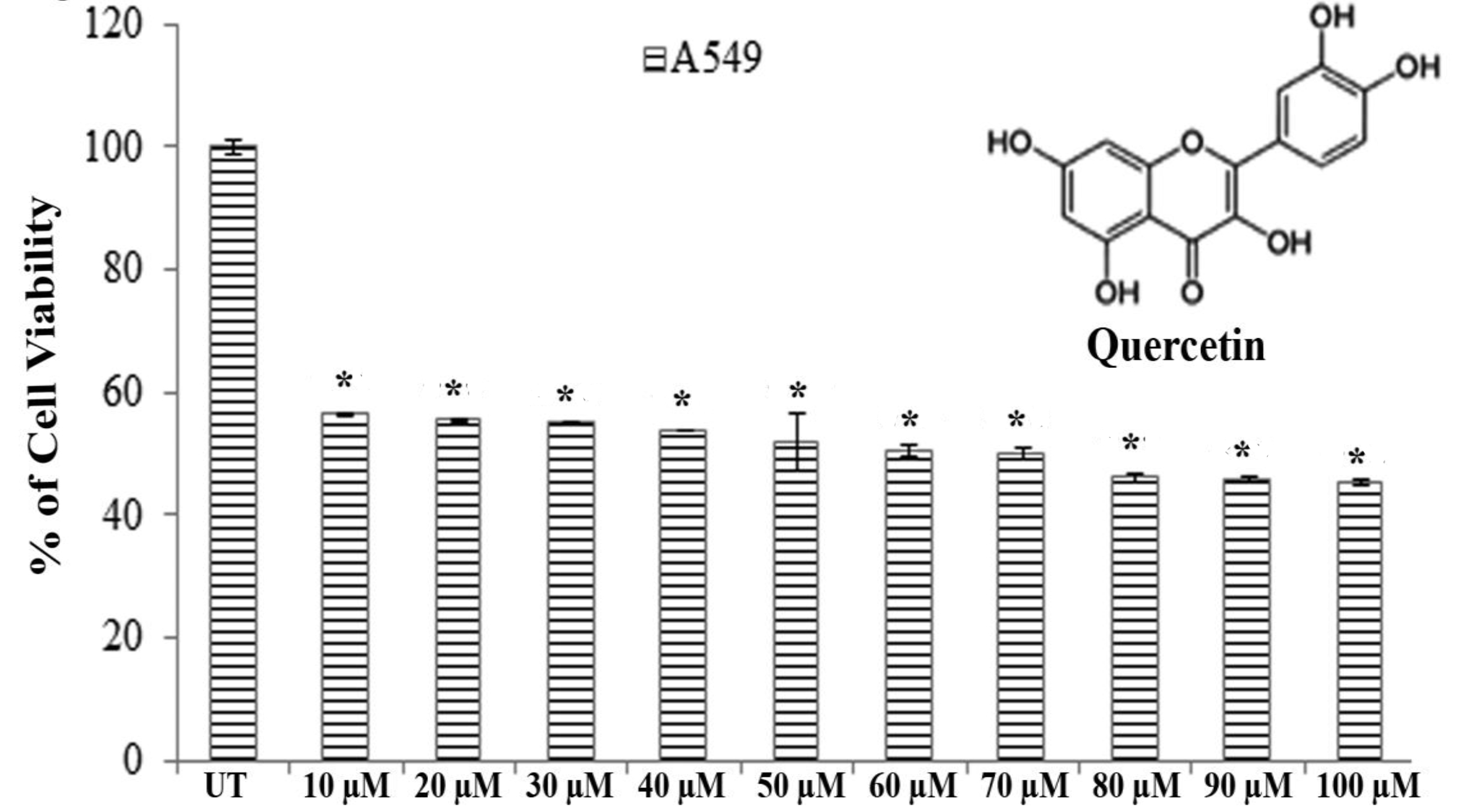
Results of MTT assays revealed that significantly greater cytotoxicity was induced in A549 cells than in H460 cells after exposure to quercetin for 24 hours. Quercetin showed an almost 50% A549 cell death at a dose of 66.5 ± 0.083 μM after 24 hours of incubation whereas H460 showed the same percent of cell death at a dose greater than 100 μM (Fig. 1).
Therefore, we selected A549 cells for further studies to evaluate the effect of quercetin.
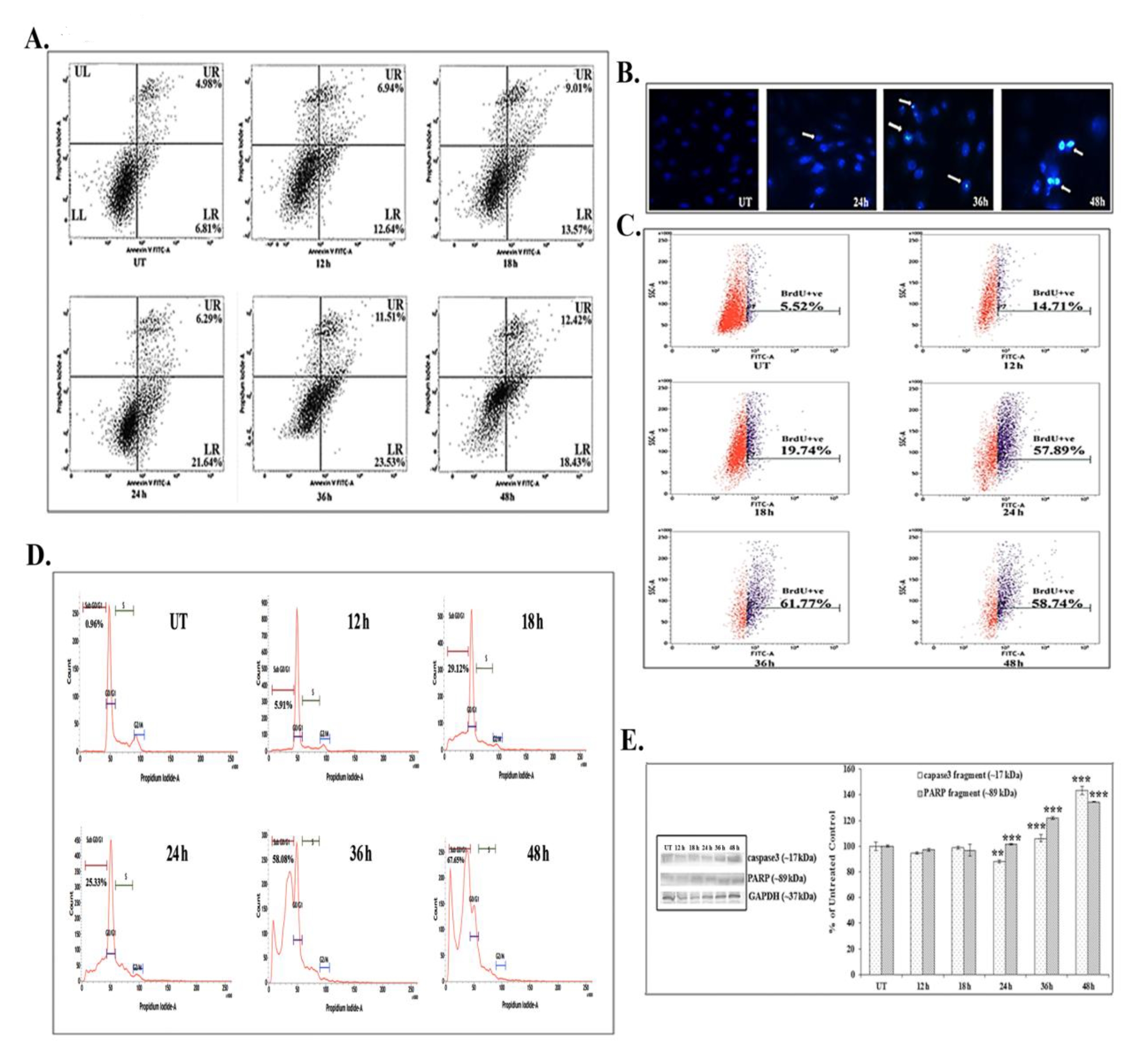
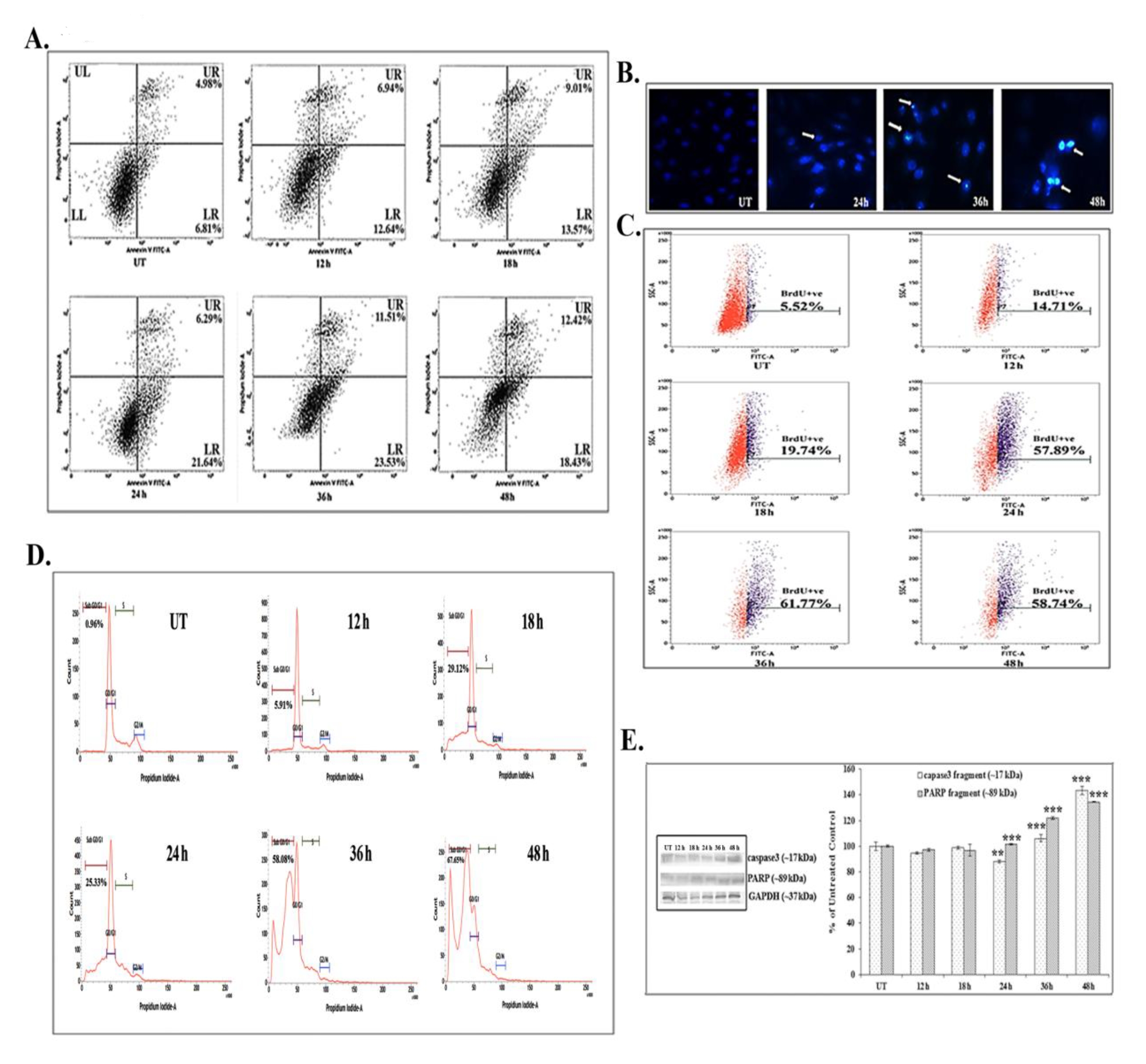
FACS analyses showed that 66 μM (IC50) of quercetin could induce phosphatidylserine exposure which is a hallmark feature of apoptosis. Quercetin at 12 – 24 hours of exposure up-regulated the AnnexinV positive and the PI negative cell populations (early state of apoptosis). However, at 24 – 48 hours of quercetin exposure, both the Annexin V and the PI positive cell populations were found to be elevated, which would indicate that the cells were at a late state of apoptosis as compared with untreated control cells (Fig. 2A).
The DAPI staining assay showed brighter fluorescence of A549 cells after 24 hours of quercetin exposure at a dose of 66 μM (Fig. 2B). The bright fluorescence occurred due to presence of nicked DNA.
A significant number of TUNEL positive cells were present after quercetin treatment (58.74% after 48 hours of treatment) as compared with untreated control cells (5.52%). The increase in the TUNEL positive cell population after quercetin exposure appeared to be time dependent in nature (Fig. 2C). The flow cytometric analysis indicated sub-diploid cell population (sub-G1) arrest in quercetin (66 μM) treated A549 cells (67.65% after 48 hours of treatment).
Quercetin induced cell cycle arrest was appeared to be occurred in a time dependent manner (Fig. 2D), as compared with untreated control cells (0.96%).
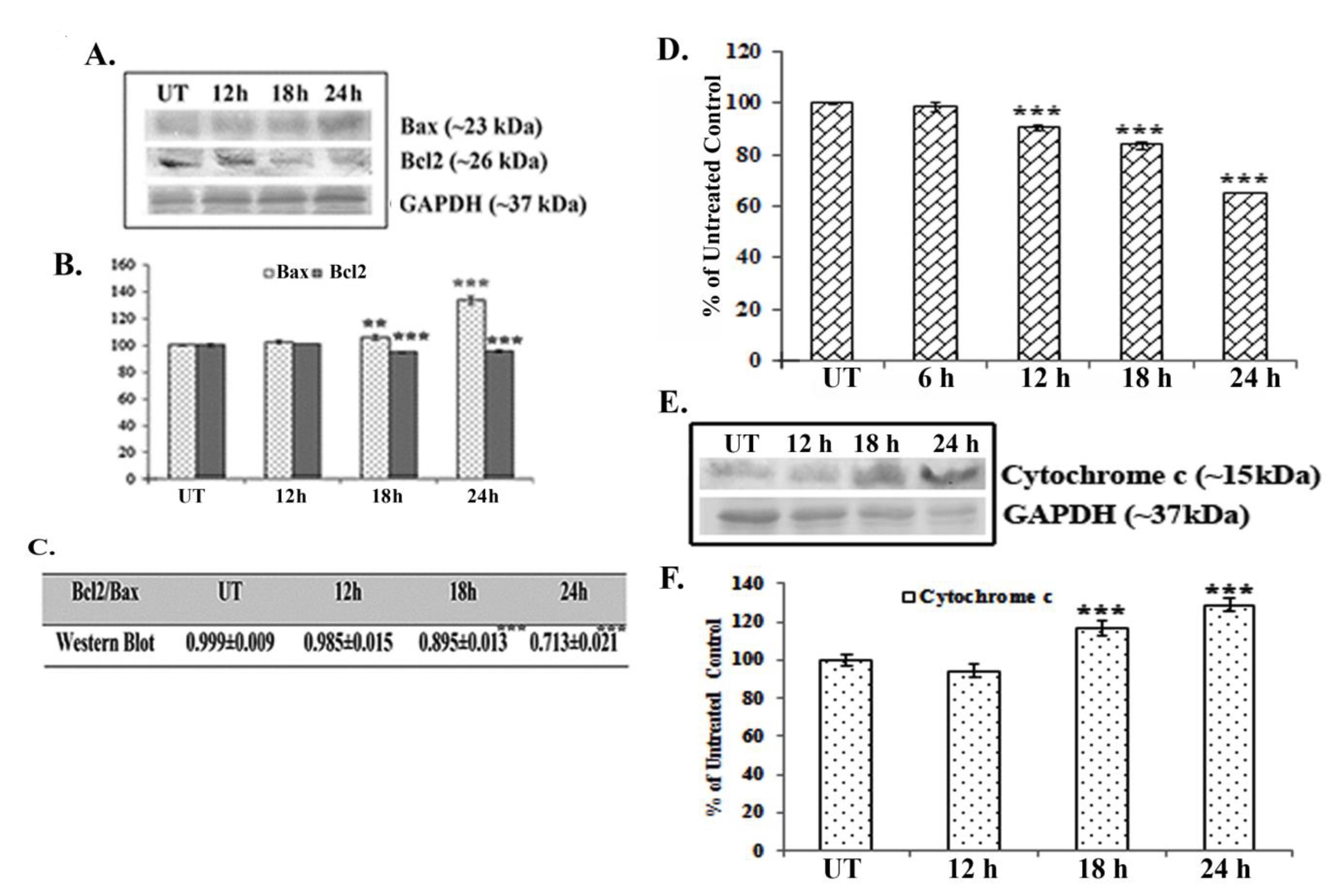
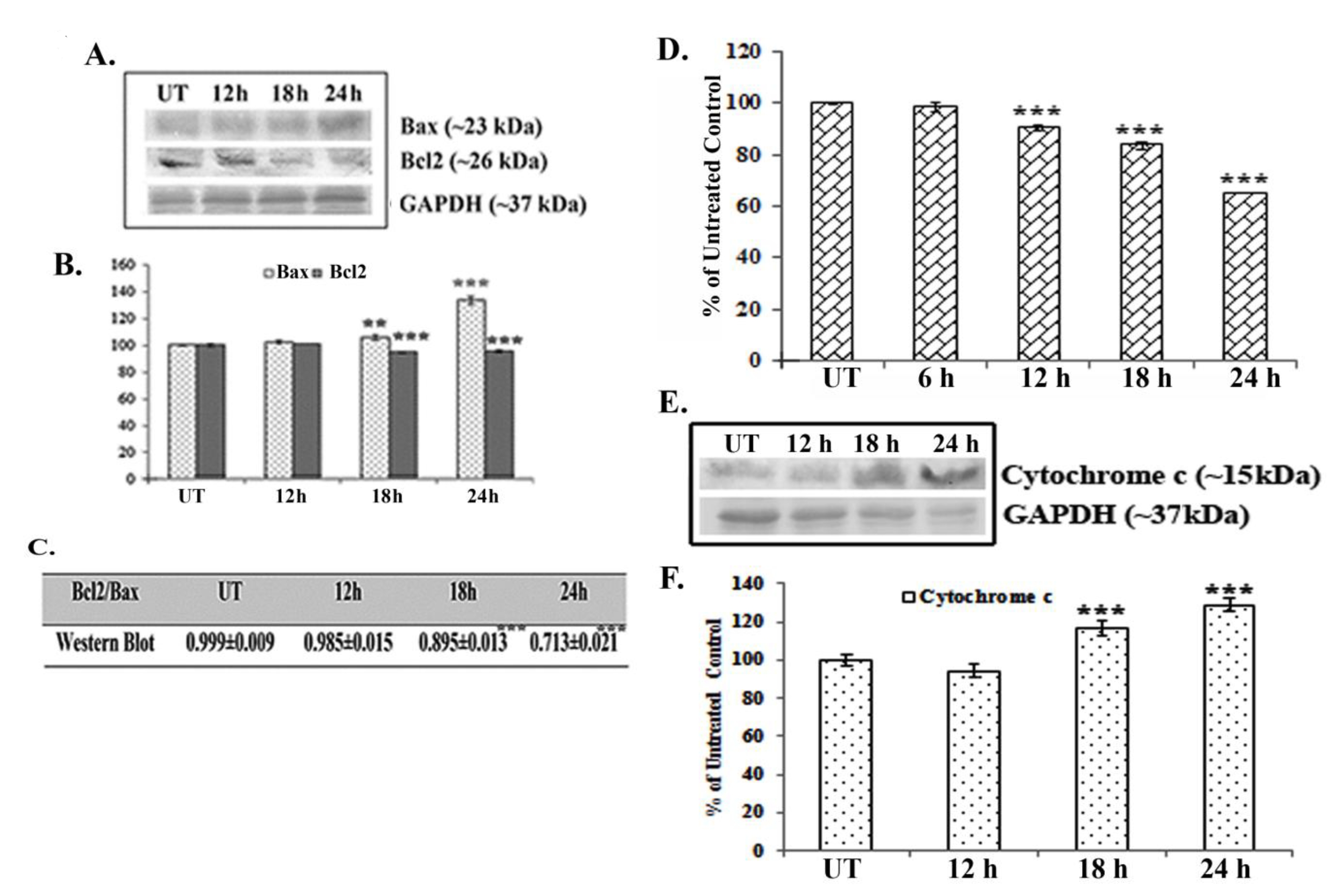
Western blot data showed a time dependent up-regulation of the caspase 3 fragment (17 kDa) and the PARP fragment (89 kDa) after treatment with 66 μM of quercetin (Fig. 2E). The up-regulation of caspase 3 appeared to be more significant after 24 hours of quercetin treatment. Analysis of western blot data showed an up-regulation of Bax and a down-regulation of Bcl2 after 12 – 24 hours of quercetin exposure at a dose of 66 μM (Fig. 3A-B). The down-regulation of the Bcl2/Bax ratio (Fig. 3C) was, thus, capable of triggering a downstream apoptotic pathway. The alteration was found to be significant after 18 hours of quercetin exposure.
Results of fluorimetry fluorimetric assay revealed that a 66-μM quercetin exposure depolarized the mitochondrial membrane’s potential significantly after 12 - 24 hours of exposure (Fig. 3D).
A crucial step of mitochondrial apoptosis is alteration in mitochondrial membrane polarization (MMP) and release of cytochrome c in cytosol, which switches on the caspase cascade [24]. Western blot data revealed an up-regulation of cytochrome-c expression in the cytosol, especially after 18 hours of quercetin exposure.
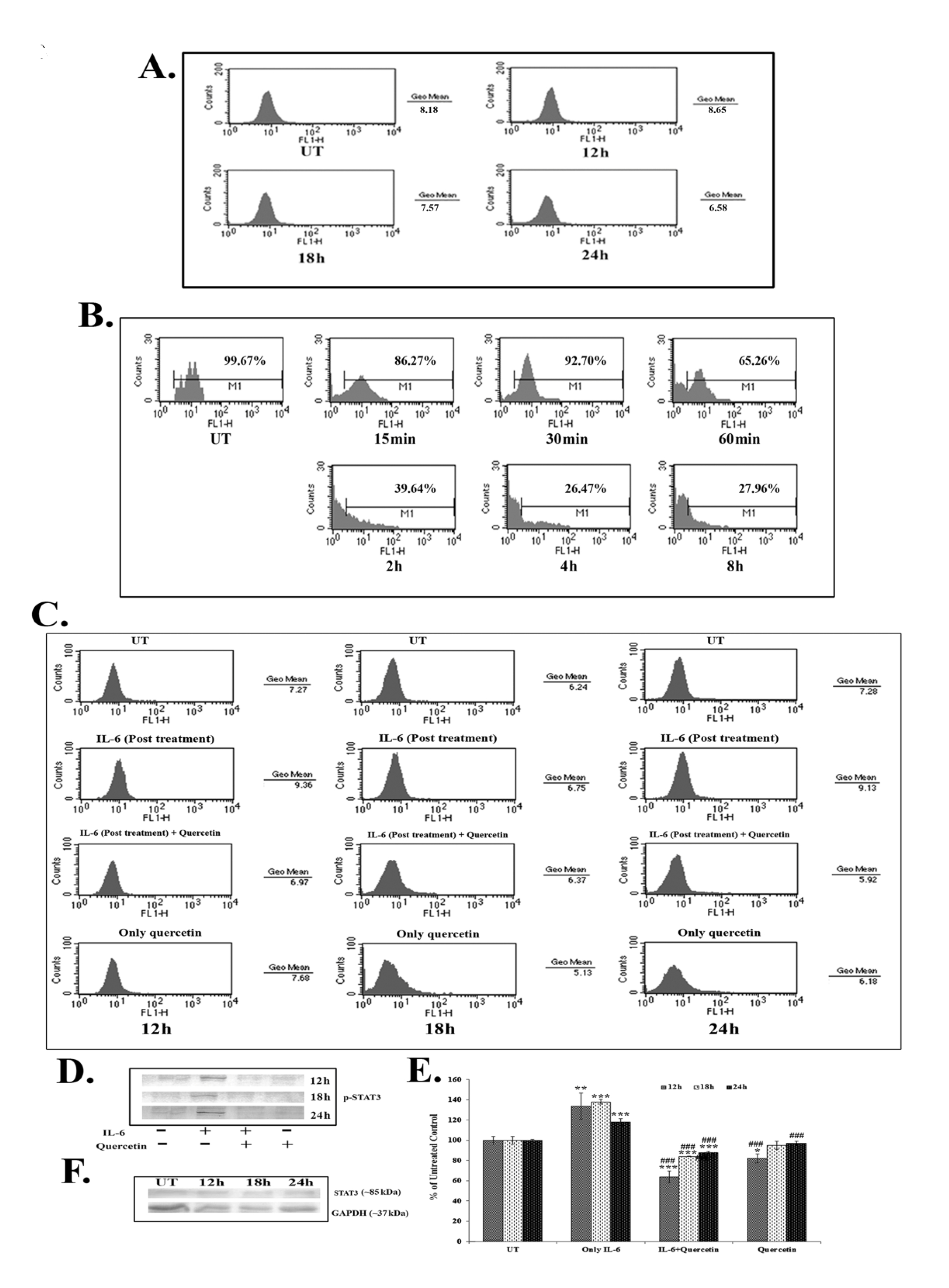
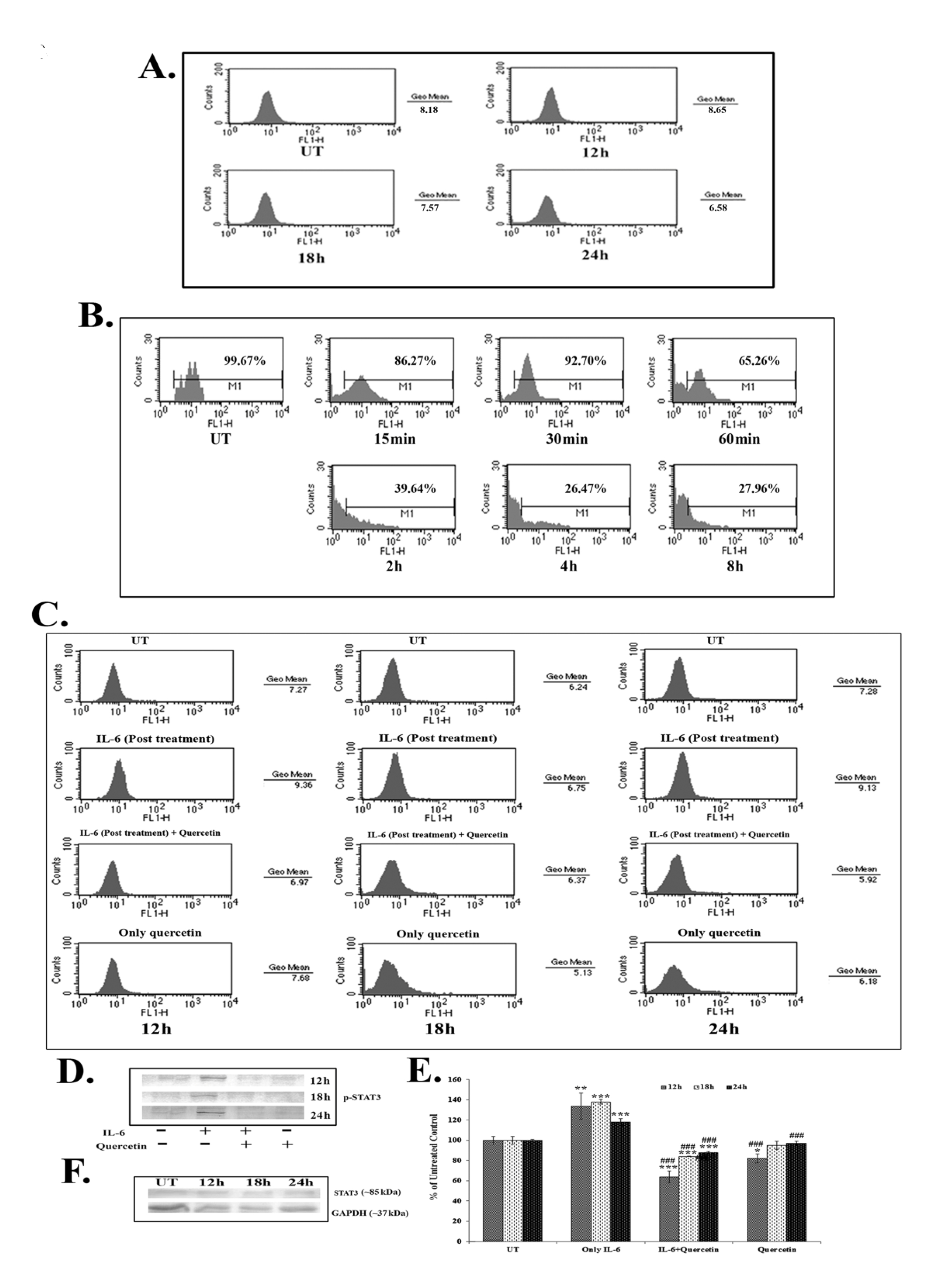
Results of the FACS assay revealed a down-regulation of IL-6 titer after 18 – 24 hours of quercetin (66 μM) exposure (Fig. 4A). IL-6 titer was found to be unchanged after quercetin treatment for 12 hours. However, from 18 hours onwards, a time dependent down-regulation of IL-6 titer was observed after quercetin treatment. NF-κB is an upstream effector molecule of IL-6 activation [15]. Results of FACS analysis revealed a lower level of nuclear translocation of NF-κB (p65 subunit) at early hours (15 – 60 minutes and 2 – 8 hours) of 66-μM quercetin treatment (Fig. 4B).
IL-6 induced p-STAT3 expression was down-regulated after 12 – 24 hours of quercetin exposure at a dose of 66 μM, as revealed by both flow cytometry (Fig. 4C) and western blot data (Fig. 4D-E). Quercetin was able to down-regulate p-STAT3 expression more significantly in the cells that received a post treatment of IL-6.
Apart from this, constitutive expression of STAT3 remained unaltered after quercetin exposure (Fig. 4F), indicating that the drug specifically down regulated only the activated STAT3 (p-STAT3) expression.
A significant number of A549 cells were destroyed when they were exposed to quercetin at doses of 10 - 100 μM. Results of MTT assay revealed that A549 cells had greater sensitivity than H460 cells upon quercetin induction. Almost a 50% A549 cell death occurred after a 66-μM quercetin treatment for 24 hours. Flow cytometry studies revealed that this cytotoxic effect of quercetin on A549 cells was apoptotic in nature, as most of the cell population showed positive response to Annexin-V and TUNEL assays. A cell cycle analysis also revealed a significant sub-diploidal (sub-G1) A549 cell population after quercetin exposure. This apoptotic effect appeared to be time dependent. For further confirmation of the efficacy of quercetin, the activities of apoptosis related proteins were analyzed by using western blots. Data showed that quercetin at a dose of 66 μM activated caspase 3, major gene related to apoptosis. Significant up-regulations of cleaved caspase 3 (17 kDa) and PARP (89 kDa) expressions, especially at late hours of quercetin treatment (24 – 48 hours), supported the involvement of caspase 3 during the apoptotic process.
Major pro- and anti-apoptotic proteins regulate cell survivability by maintaining a proper balance in the cell [16]. Therefore, modulations of major pro- and anti-apoptotic proteins are thought to be involved in drug induced apoptotic pathways [25]. Consistent with this, western blot data showed an up-regulation of the major pro-apoptotic protein Bax and a down-regulation of the major anti-apoptotic protein Bcl2 after quercetin exposure in A549 cells. Therefore, the down-regulation in the Bcl2/Bax ratio was found to be significant at 18 – 24 hours of quercetin exposure. Elmore [26] in his earlier studies reported that mitochondrial membrane depolarization during apoptosis might be responsible for any drug induced Bcl2/Bax imbalance. In this context, a fluorimetry study was done to measure the mitochondrial membrane’s potential after staining with Rhodamine 123. Results indicated that mitochondrial membrane depolarization occurred at 12 – 24 hours of quercetin exposure, which would indicate the quercetin induced A549 cell apoptosis to be a mitochondria mediated one.
STAT3, a member of the STAT family of transcription factors is a key regulator involved in cancer related inflammation [12, 27]. STAT3 is frequently deregulated during lung cancer [13]. Activation of STAT3 results in the expression of downstream genes, such as Bcl2, that control cell proliferation, invasion and survivability [15, 16]. As quercetin was found to modulate the expression of Bcl2, the effect of it on STAT3, a major upstream effector molecule of Bcl2 [15], was analyzed. IL-6, a major cancer related inflammatory cytokine, regulates STAT3 activation, and its up-regulation in lung cancer is thought to promote lung tumorigenesis [11]. Therefore, the study was designed to establish the efficacy of quercetin, if any, in modulating the IL-6 mediated STAT3 activation. Quercetin was found to be able to down-regulate IL-6 mediated STAT3 activation (p-STAT3) after 18 – 24 hours of A549 cells exposure to it, as revealed from the western blot and the FACS data. The efficacy of quercetin was found to be very large in inhibiting IL-6 induced STAT3 activation, especially in those cells that had been post treated with IL-6. This led us to hypothesize that secretion of IL-6 from the surrounding microenvironment or neighboring cells would not be able to raise the activation of STAT3 after treatment with quercetin at the preferred dose. As IL-6 induced STAT3 activation was found to be down-regulated after quercetin exposure, a subsequent FACS analysis was done to measure IL-6 titer upon quercetin exposure. A flow cytometric analysis revealed decreased IL-6 titer after 18 – 24 hours of quercetin exposure in A549 cells.
NF-κB, a major upstream transcription factor of IL-6 [10], has been reported to be activated after translocation to a nucleus from the cytoplasm and plays a major role in tumorigenesis [28]. Earlier, Yamamoto and Gaynor [29] suggested NF-κB as a potential target in the treatment of inflammation induced cancer. The present study highlighted that quercetin could modulate IL-6 induced STAT3 activation by decreasing cellular titer of IL-6 in A549 cells. Therefore, the study was intended to reveal the modulatory role, if any, of quercetin on NF-κB activation. A nuclear translocation study after the FACS analysis showed a down-regulation of NF-κB (p65 subunit) at early hours of quercetin exposure (from 15 minute to 6 hours). NF-κB, on the other hand, is a major upstream transcription factor of Bcl2 [14]. Thus, down-regulation of quercetin induced NF-κB activation might play a dual function in A549 cells, creating positive modulation of both IL-6 and Bcl2, which would lead the cell into apoptosis. This observation indicated that, consistent with the results reported by earlier workers, the reduction of the Bcl2 protein level caused by quercetin might have occurred as a result of the inhibition of the IL-6 induced STAT3 signaling and by the down-regulation of NF-κB activity [14, 15]. An early down-regulation of quercetin induced NF-κB activity is thought to have played a vital role, as this, in turn, could modulate both the expressions of IL-6 and Bcl2 in A549 cells. Down-regulation of IL-6 titer helped in lowering the inflammatory responses of A549 cells. Simultaneously, all these factors could, therefore, contribute to the modulation of the Bcl2/ Bax imbalance, which was thought to promote quercetin induced mitochondria mediated apoptosis in A549 cells.
In conclusion, targeted blockage of the oncogenic functions of NF-κB and IL-6/STAT3 signaling by quercetin represents a new strategy for therapeutic application against NSCLC with constitutive IL6/STAT3 activation for uncontrolled cell proliferation through immune surveillance. As such, inhibition of the IL-6/STAT3 signaling pathway, particularly by modulating NF-κB activation, in turn, leads to a Bcl2/Bax imbalance, which can direct the cell itself to enter into self-destruction by triggering apoptosis. For an understanding of the mechanism of action of quercetin against the NSCLC cell line, observation of its effects on the A549 cell line may provide valuable information for its potential application in NSCLC prevention and therapy. Further, quercetin was found to be a strong agent that induced mitochondrial mediated apoptosis in A549 cells through down-regulation of the IL-6/STAT3 pathway. NF- κB, a major upstream effector of IL-6 and Bcl2, was also found to be down-regulated at early hours by quercetin, resulting in a decrease in the IL-6 titer. Thus, down-regulation of IL-6 also deactivated STAT3 (p-STAT3) expression, modulating the Bcl2 response as NF-kB and STAT3 had been reported to be major upstream effectors of Bcl2. Altered Bcl2 response, thus, induced mitochondrial depolarization that led the cell ultimately into mitochondriamediated apoptosis. Overall results provide a strong indication that quercetin is a potentially effective therapeutic drug for use against NSCLC.