In this work, nanoporous carbons (NPCs) were prepared by the self-assembly of polymeric carbon precursors and block copolymer template in the presence of tetraethyl orthosilicate and colloidal silica. The NPCs’ pore structures and total pore volumes were analyzed by reference to N2/77 K adsorption isotherms. The porosity and elemental mercury adsorption of NPCs were increased by activation with carbon dioxide. It could be resulted that elemental mercury adsorption ability of NPCs depended on their specific surface area and micropore fraction.
Coal-fired power plants are one of the primary sources of mercury emissions. In the United States, nearly one third of the 150 tons of mercury emitted comes from coal-fired power plants. Coal is a major energy resource which is mainly burnt to produce electricity. As is known, it is not a clean fuel. Various types of pollutants, such as SOx, NOx and mercury, are released into the atmosphere during the burning of coal. Concerns over mercury emissions have increased due to the high toxicity, volatility, and bioaccumulation of mercury in the environment and due to the neurological health impact of mercury. According to the Global Mercury Assessment Report, coal-fired power plants are the primary source of the anthropogenic emissions of mercury into the atmosphere [1-8].
Porous carbon materials have gained much attention due to their high specific surface area, good thermal stability, tunable porosity, chemical inertness, and biocompatibility. These features contribute to their high performance in various applications, such as the adsorption of dye molecules, which have become one of the most serious air/water pollutants [9-14]. Activated carbon, owing to its large specific surface area and high pore volume, is frequently used as an adsorbent [15-21]. However, its widespread use is restricted due to the micropore size, which limits the transfer of bulky dye molecules from the surface of active carbon into the pores [11,22,23]. Therefore, ordered mesoporous carbons (OMCs) with uniformly large pores and a high surface area are in great demand.
The versatility of carbon-based nanomaterials makes them very appealing for a number of applications, including catalysis, adsorption, separation, and energy storage. Many of these applications require carbons with well-developed micro- and mesoporosity. While microporosity can be easily created by the carbonization of certain natural materials or through the use of CO2, steam or KOH as an activating agent [24,25], the formation of mesopores is more challenging.
There have been extensive developments related to the synthesis of OMCs [26] over the last decade. Initial reports were based on the hard-templating method, in which the pore geometry and size of the mesopore structure can be tuned by the choice of template materials. However, a major drawback of this synthesis route is the large number of preparation steps correand, in some cases, the fairly limited control of the mesopore diameters. The recently reported soft-templating approach [26] greatly simplified the preparation of OMCs. The use of triblock copolymers and thermosetting carbon precursors permits the direct formation of polymer–polymer composites, which, when subjected to a controlled thermal treatment, can be converted into mesoporous polymers and finally to mesoporous carbons. This synthesis route allows more freedom in the design of the symmetry and size of mesopores through simple changes in the synthesis conditions and/or the triblock copolymers used as soft templates. Though several synthetic pathways have been reported to date for the synthesis of mesoporous materials, a general one-step synthetic strategy to prepare crystalline nanoporous materials is still desired [27,28].
In this study, a direct, one-step synthetic protocol which can be used to synthesize nanoporous carbons (NPCs) is reported. The essence of this method is its direct use of the self-assembly of block copolymers as templates for the generation of porous metal-carbon structures, without the extra step of generating templating silica structures. Mesoporous carbons were synthesized using a tri-block copolymer (structure-directing agent), a mixture of resorcinol and formaldehyde (carbon source) under a mild acidic condition, after which the efficiency of elemental mercury adsorption given the activation condition was evaluated.
Approximately 1.25 g of resorcinol (Aldrich, USA) and 1.25 g of Pluronic F127 (Aldrich) were dissolved in deionized water and ethanol. The weight ratio of water to ethanol was fixed at 5.5:10. After stirring for about 10-15 min, the reaction mixture was supplied with 1.1 mL of 37% HCl with continual stirring for an additional 30 min. Next, 1.25 mL of formaldehyde and 1.87 mL of tetraethyl orthosilicate were added to the synthesis mixture. The resulting solution turned milky after 2-3 h, and after an additional 30 min of stirring, the aqueous and solid phases were allowed to separate. The polymer-containing bottom layer was spread on a Petri dish and left under the fume hood overnight. Then, the sample was aged at 100℃ for 24 h. A thermal treatment was performed in a nitrogen atmosphere in a tube furnace using a heating rate of 2℃/min up to 180℃. The sample was kept at this temperature for 5 h and then heated at a rate of 2℃/ min up to 400℃, and then at a rate of 5℃/min up to 850℃. Finally, the sample was kept at 850℃ for 2 h.
For activation, the samples were heated in a tube furnace in a nitrogen atmosphere at a heating rate of 5℃/min up to the activation temperature. After reaching this temperature, an activating gas was introduced into the tube furnace (80 cm3/min) for 1 h, with switching back to nitrogen to prevent further activation during the cool-down process. The activation temperature was varied at 600℃, 700℃, 800℃, and 900℃.
The N2 adsorption isotherms were measured using a Belsorp Max (BEL Japan) at 77 K. The samples were degassed at 573 K for 12 h to obtain a residual pressure of less than 10-6 torr. The amount of N2 adsorbed onto the samples was used to calculate the specific surface area by means of the Brunauer-Emmett-Teller (BET) equation [29]. The total pore volume was estimated to be the liquid volume of the N2 at a relative pressure of approximately 0.995. The micropore volume was calculated using the Dubinin-Radushkevitch (D-R) equation [30].
2.3. Elemental mercury removal efficiency
The reactor, with a 12.7 mm diameter and made of quartz, was placed inside a temperature-controllable tubular furnace which was heated to a temperature of 70℃. For every experiment, 1.0 g of sample materials were loaded and packed inside a quartz tube. A carrier gas was fed into the adsorption apparatus at a flow rate of 100 mL/min. Elemental mercury gas was generated from elemental mercury permeation tubes (Dynacalibrator® Model 150; VICI Metronics Inc., USA), and the concentration of the elemental mercury gas was maintained at 700 ppm during the experimental process. Measurements of both the inlet and outlet concentrations of the elemental mercury were done using a mercury analyzer (VM-3000; Mercury Instruments, Germany), and sulfur was used to capture the elemental mercury from the effluent gas.
Fig. 1 shows high-resolution scanning electron microscope images of the NPCs as a function of the activation temperature. An obvious hexagonal arrangement of the mesopores is observed for all samples, suggesting that an ordered mesostructure is maintained.
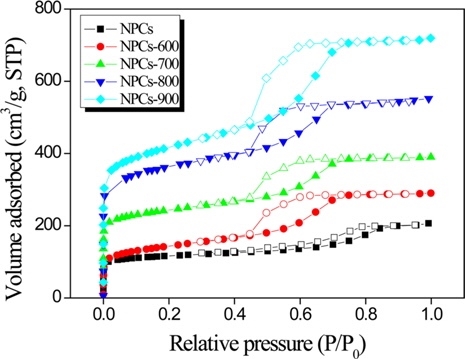
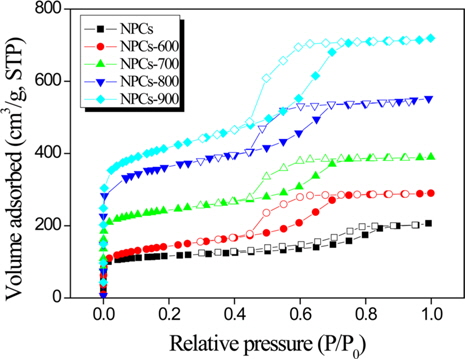
Good understanding of the porosity and the specific surface area of an adsorbent can be achieved by constructing an adsorption isotherm of N2. Fig. 2 shows the N2 adsorption isotherms of the NPCs as a function of the activation temperature. All carbons show type IV adsorption isotherms, indicating the presence of a well-developed mesoporous structure. The capillary condensation steps are nearly perpendicular to the relative pressure axis, suggesting high uniformity of the mesopores. The isotherm has a sharp slope at a relative pressure of 0 to 0.1, which can be attributed to the presence of micropores, and a small slope at a relative pressure of 0.4 to 0.8, which indicates a broad pore-size distribution in the mesopore range. This is identified as a slow rate of increase in N2 uptake at a low relative pressure, correand sponding to monolayer-multilayer adsorption on the pore walls. Additionally, the capillary condensation step became slightly broader, implying some minor deterioration of the mesopore structure.
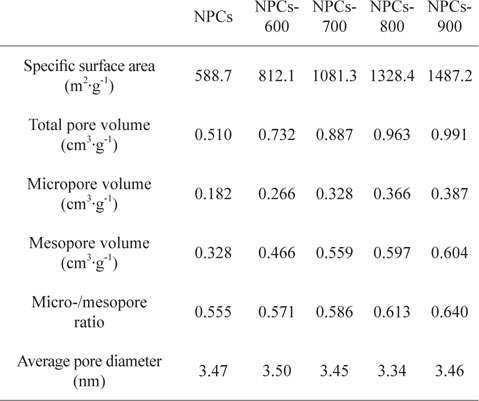
Table 1 lists detailed information of the textural properties of the samples from the N2/77 K adsorption isotherms. The NPC sample had a specific surface area of 589 m2/g, a total pore volume of 0.510 cm2/g and a micropore volume of 0.328 cm2/g. The activated samples manifested an increasing specific surface area and increasing total and micropore volumes as the activation temperature increased. This suggests that higher CO2 activation temperatures definitely alter both types of pores, as reflected by the high surface area and total pore volume.
[Table 1.] Textural properties of the NPCs as a function of the activation temperature
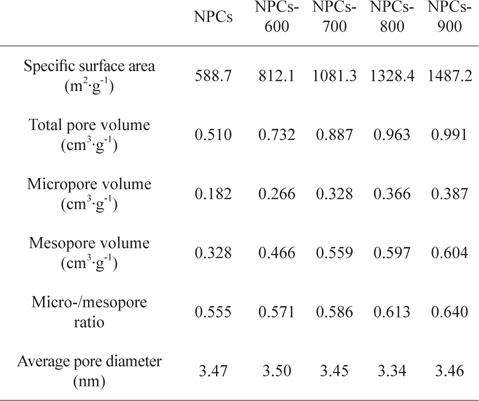
Textural properties of the NPCs as a function of the activation temperature
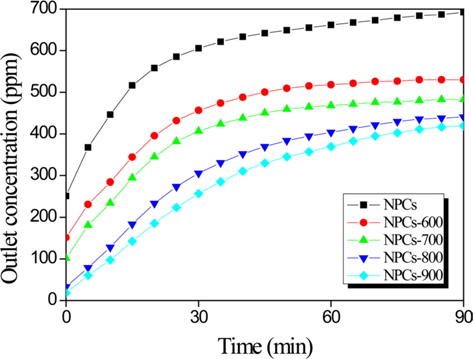
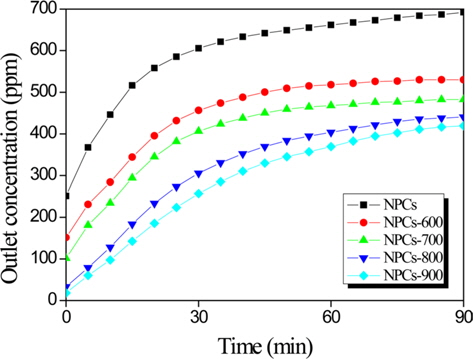
Fig. 3 shows the elemental mercury removal efficiency of the ordered NPCs. All tests were conducted at 70℃ for 90 min in an elemental mercury adsorption apparatus. Normally, gas adsorption depends strongly on the specific surface area and pore volume of the adsorbents. The NPCs showed only a low elemental mercury adsorption rate of 8 ppm at 90 min. Meanwhile, the elemental mercury removal efficiency of the CO2-activated NPCs was higher than that of the NPCs due to the clearly higher specific surface area and micro-/mesopore ratio. In conclusion, the optimal materials for mercury vapor adsorption possess the following attributes: 1) a high-specific surface area, and 2) a high-micropore fraction.
In this work, we investigated the synthesis of phenolic resin- based carbons by a combination of templating followed by post-synthesis activation in an effort to enhance the elemental mercury adsorption behaviors. The results of this study indicate that CO2 activation offers high elemental mercury removal efficiency of ordered NPCs. We can conclude this work by stating that mercury vapor adsorptions rates can be optimized in terms of the specific surface area and micropore fraction.