Dieckol is a polyphenol compound isolated from brown algae that has anti-oxidant, anti-inflammatory, and anti-tumor activity. We examined the anti-angiogenic effects of dieckol in endothelial cells under hypoxic conditions. Treatment with CoCl2, a hypoxic mimetic agent, increased proliferation, adhesion, migration, and tube formation in HUVECs, as well as vessel sprouting in rat aortic rings, which correlated well with increased expression of hypoxia-inducible factor 1-alpha (HIF1α) and β1-integrin. Dieckol suppressed CoCl2-induced adhesion, migration, and tube formation in HUVECs and vessel sprouting in rat aortic rings. Dieckol treatment decreased CoCl2-induced overexpression of HIF1α and its downstream signaling molecules, including β1-integrin/Fak, Akt/eNOS, and p38 MAPK. These results suggest that dieckol is a novel angiogenesis inhibitor and a potential treatment for angiogenesis-dependent diseases in humans, such as malignant tumors.
Angiogenesis is the physiological process in which new blood vessels are generated from pre-existing vessels, and it is involved in healing and reproductive processes, including wound healing, female reproduction, embryonic development, organ formation, and tissue remodeling. Angiogenesis is strictly controlled by a wide range of regulators (Bussolino et al., 1997). Hypoxia-inducible factor 1α, HIF1 α, is a key regulator of angiogenesis that promotes the expression of a broad range of genes that respond to low oxygen concentrations, and which induces the formation of new blood vessels. Therefore, hypoxia-induced angiogenesis has become an attractive therapeutic strategy for the treatment of many human diseases, including cancer, ischemic heart disease, peripheral artery disease, and neovascular eye diseases, as well as a strategy to accelerate wound healing (Krock et al., 2011). However, the mechanisms involved in hypoxia-induced angiogenesis and the best manner in which to exploit it for the treatment of human diseases are not well understood and still under active investigation.
Angiogenesis is a complex process that includes endothelial proliferation, migration, degradation of the extracellular matrix, tube formation, and sprouting of new capillary branches, which depend on coordinated signaling by growth factors and cell adhesion receptors. Integrin is the principle adhesion receptor used by endothelial cells to interact with the extracellular microenvironment, and integrin-mediated interactions play a critical role in regulating proliferation, migration, and survival of endothelial cells. Many studies have focused on the role of integrin family members αVβ3 and αVβ5, because earlier studies using antagonists or neutralizing antibodies for these proteins reported dramatic inhibition of angiogenesis (Brooks et al., 1994; Friedlander et al., 1995). However, mice lacking αV, β3, or β5, which are the constituent proteins of αVβ3 and αVβ5, do not show obvious angiogenic defects (Bader et al., 1998; Francis et al., 2002), raising the possibility that other integrin members control angiogenesis. Accumulating evidence suggests that β1-integrin is involved in angiogenesis. β1-Integrin null mice show reduced vasculature and irregular vessel formation (Bloch et al., 1997). Tumor-associated vessels have been reported to overexpress fibronectin and β1-integrin, and treatment with function-blocking antibodies and inhibitory molecules against β1-integrin inhibits angiogenesis in response to tumor growth factors (Mettouchi and Meneguzzi, 2006). In addition, several endogenous inhibitors of angiogenesis have been reported to directly bind to β1-integrin. Endorepellin and endostatin, which are proteolytic fragments of the membrane protein perlecan or collagen XVIII, exhibit anti-angiogenic activity through binding to β1 integrin (Sudhakar et al., 2003; Bix et al., 2004), strongly suggesting a major role for β1-integrin in angiogenesis. Several recent studies have also reported that expression of fibronectin and β1-integrin are increased under hypoxic conditions, and this overexpression regulates the proliferation and migration of progenitor cells and stem cells, although the related signaling mechanisms have not been defined (Irigoyen et al., 2008). Therefore, in this study we focused on the role of β1-integrin in hypoxia-induced angiogenesis in human umbilical vein endothelial cells (HUVECs), and particularly on the anti-angiogenic effects of dieckol, which was identified as an inhibitor of β1-integrin signaling in our recent screening study (Park and Jeon, 2012). Dieckol is a phlorotannin isolated from brown algae that has recently been reported to exhibit various biological activities in multiple cell types (Heo et al., 2009; Lee et al., 2010). However, its biological and biochemical effects in endothelial cells have not been reported.
Human umbilical vein endothelial cells (HUVECs) were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in M199 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 20% fetal bovine serum (FBS), 100 units/mL penicillin, 100 μg/mL streptomycin, 3 ng/mL basic fibroblast growth factor (Upstate Biotechnology, NY), and 5 units/mL heparin. The cells were grown in 5% CO2 in air at 37°C. For the experiments, cells were detached with trypsin-EDTA. Matrigel was obtained from BD Biosciences (St Louis, MO, USA). Mouse monoclonal anti-human β1-integrin antibody was from Cell Signaling Technology (Beverly, MA, USA). Antibodies against Fak, Y397 Fak and primary antibodies rabbit anti-SAPK, anti-pSAPK, 306and beta-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against Akt, pAkt, eNOS, and p38 MAPK were purchased Cell Signaling Technology (Beverly, MA, USA). Dieckol was prepared as reported by Lee et al. (2010). Dieckol did not affect viability of HUVECs at the concentrations of below 50 μM (Data not shown). The treatment of HUVECs with non-cytotoxic 25 μM dieckol induced maximal decrease in the Cocl2-induced cell adhesion and migration (Data not shown). Therefore, 25 μM of dieckol was used in all the experiments.
>
Cell viability and cell proliferation assay
HUVECs were cultured for 4 passages. HUVECs were then trypsinized, plated at 6×103 cells/well on 12-well tissue culture plates (Falcon, USA) and continuously cultured in 3 mL M199 containing 1% FBS. When the cells were 80–90% confluent, the culture medium was replaced with serum-free M199 for 12 h, after which the cultures were divided into 2 groups: with or without 25 μM dieckol. The viability of cells after dieckol treatment was assessed by the MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. HUVEC morphology was assessed with phase-contrast microscopy and cell counts were also calculated by hemocytometry.
HUVECs were incubated in the presence or absence of 100 μM CoCl2 and 25 μM dieckol for 24 h. The cells were harvested and resuspended in culture medium. The cells were then transferred to a 24-well plate that was precoated with fibronectin (25 μg/mL). After incubation for 1 h at 37°C, the medium was discarded and washed with PBS to remove the non-adherent cells. Attached cells were photographed and quantified.
HUVEC migratory function, which is essential for angiogenesis, was examined using a modified Boyden chamber technique. A 24-well Transwell apparatus (Costar) was used, with each well containing a 6.5 mm polycarbonate membrane with 8 μm pores that was coated with type I collagen (Sigma-Aldrich, St. Louis, MO, USA). HUVECs (4×104) were placed on the membrane, and the chamber was immersed in a 24-well plate that was filled with growth factor-free M199 culture media with or without 25 μM dieckol. After incubation for 24 h, the membrane was washed briefly with PBS and the upper side of the membrane was wiped gently with a cotton ball, after which it was removed and stained with hematoxylin and eosin (H&E). The magnitude of HUVEC migration was evaluated by counting the migrated cells in 4 random high-power (100×) microscope fields.
>
Matrigel tube formation assay
A Matrigel tube formation assay was performed to assess the ability of HUVECs to form endothelial cell vascular structures, because this ability is believed to be important in the formation of new vessels (Rafii and Lyden, 2003). Briefly, 250 μL of growth factor-reduced Matrigel (Becton Dickinson) was pipetted into a 16-mm diameter tissue culture well and polymerized for 30 min at 37 °C. HUVECs incubated in M199 medium with 1% FBS for 12 h were harvested after trypsin treatment and suspended in M199 medium with 1% FBS. Next, 25 μM dieckol was added to the cells for 30 min at room temperature, after which they were seeded and plated onto a layer of Matrigel at a density of 2×105 cells/well, followed by the addition of 100 μM CoCl2. After 16 h, the cultures were photographed. The area covered by the tube network was determined using an optical imaging technique, in which pictures of the tubes were scanned in Adobe Photoshop and analyzed with Image-Pro® Plus 4.5 (Media CyberMetics, Inc.).
As described previously (Nicosia and Ottinetti, 1990), aortas were harvested from Sprague-Dawley rats at 6 weeks of age. Plates (48-well) were coated with 120 μL of Matrigel. After gelling, the rings were placed in the 48-well plates and sealed in place with an overlay of 50 μL of Matrigel. CoCl2 with or without dieckol was added to the wells in 200 μL of human endothelial serum-free medium (Invitrogen). Medium alone was added to the negative control cells. On day 4, cells were fixed and stained with Diff-Quick. Each treatment was assayed six times.
HUVEC cells were treated with 100 μM CoCl2 or 25 μM dieckol and then the cells were homogenized in RIPA buffer. The lysates were centrifuged at 10,000×g for 15 min, supernatant was subjected to SDS-PAGE and transferred to PVDF. The levels of protein in each sample were determined by immunoblotting with antibodies.
All data are presented as mean ± SEM. The significance of differences between the means were analyzed by Student’s ttest. All p-values of less than 0.05 were considered significant.
>
Dieckol inhibits CoCl2-induced proliferation, adhesion, and migration of HUVECs
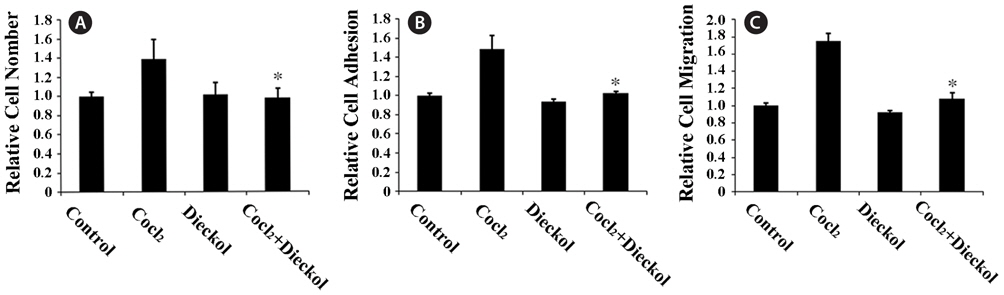
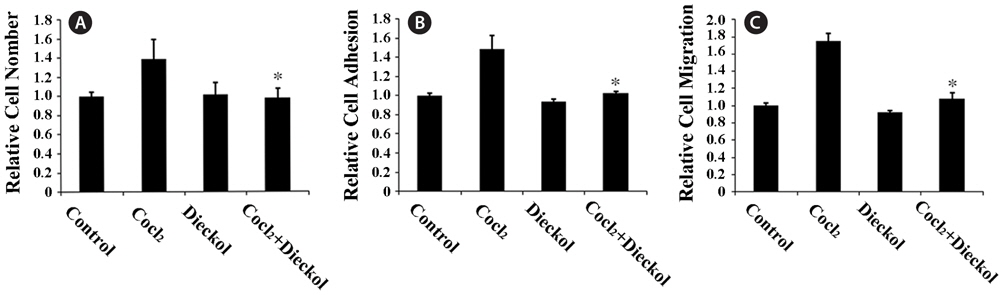
To determine the anti-angiogenic activity of dieckol, we first examined hypoxia-induced proliferation, adhesion, and migration of HUVECs. HUVECs were treated with 100 μM of the hypoxic mimetic agent CoCl2 for 24 h as described previously (Zagzag et al., 2003; Newcomb et al., 2005) in the absence or presence of 25 μM dieckol. CoCl2 treatment increased the proliferation of HUVECs by 37% compared to PBS-treated control cells, and dieckol significantly inhibited the CoCl2-induced proliferation (Fig. 1A). This inhibitory effect of dieckol was not due to the cytotoxicity of dieckol to endothelial cells, because 25 μM dieckol treatment had no effect on normal growth of HUVECs. In addition, CoCl2 treatment increased the attachment and migration of HUVECs onto fibronectin (Fig. 1B and 1C). HUVECs were preincubated in the absence or presence of 100 μM CoCl2 and 25 μM dieckol for 24 h, and then transferred to the fibronectin-coated plates for the adhesion assay or scratched by a pipette for the wound-healing migration assay. After incubation for 1 h or 16 h, attached cells were quantified and the distances of migrating cells to the wound origin were measured, respectively (Fig. 1B and 1C). Dieckol alone had no significant effect on basal attachment and migration of cells. Treatment of HUVECs with CoCl2 induced increases of approximately 48% and 75% in attachment and migration, respectively, in comparison to the control cells, whereas simultaneous dieckol treatment almost completely abolished these effects. These results suggest that dieckol regulates hypoxia-induced angiogenic responses that depend on the extracellular adhesion of HUVECs.
>
Dieckol inhibits CoCl2-induced capillary-like tube formation in HUVECs and aortic ring sprouting
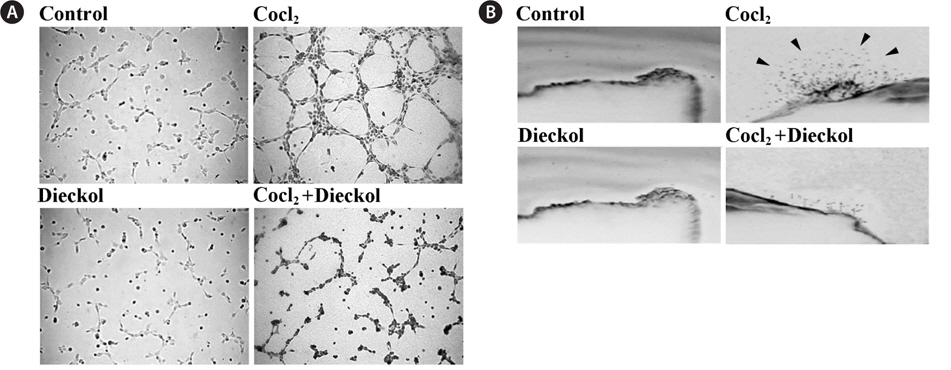
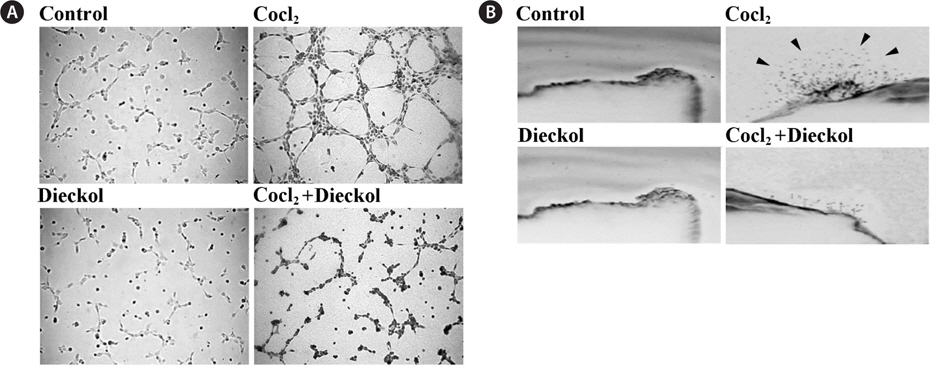
We measured the effect of CoCl2 and dieckol on the ability of HUVECs to form capillary-like structures on Matrigel. As shown in Fig. 2A, 16 h incubation with CoCl2 promoted the formation of elongated and robust capillary-like tube structures, and the number of tubes was much greater than that of the control cells; CoCl2 treatment induced an increase of approximately 2-fold in comparison to the PBS-treated control cells. CoCl2-induced tube formation in HUVECs was inhibited by treatment with 25 μM dieckol. In addition, the sprouting of vessels from excised aortic rings was investigated to determine whether 25 μM dieckol inhibits CoCl2-induced angiogenesis ex vivo (Fig. 2B). We found that 100 μM CoCl2 stimulated capillary sprouting from rat aortic rings, whereas dieckol treatment significantly attenuated the CoCl2-induced vessel sprouting. Taken together, these results indicate that dieckol treatment regulates major processes involved in hypoxia- induced angiogenesis, including endothelial cell proliferation, adhesion, migration, and tube formation, as well as vessel sprouting, which suggests that dieckol influences HIF1α signaling to regulate hypoxia-induced angiogenesis in endothelial cells.
>
Dieckol inhibits CoCl2-induced expression of HIF1α and downstream signaling molecules in HUVECs
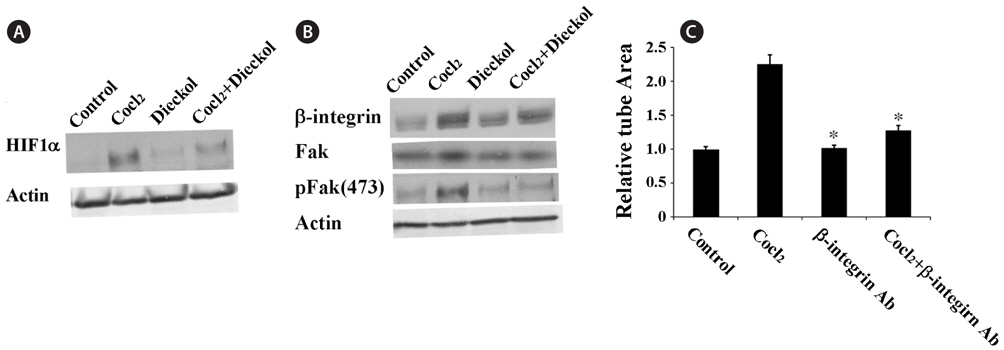
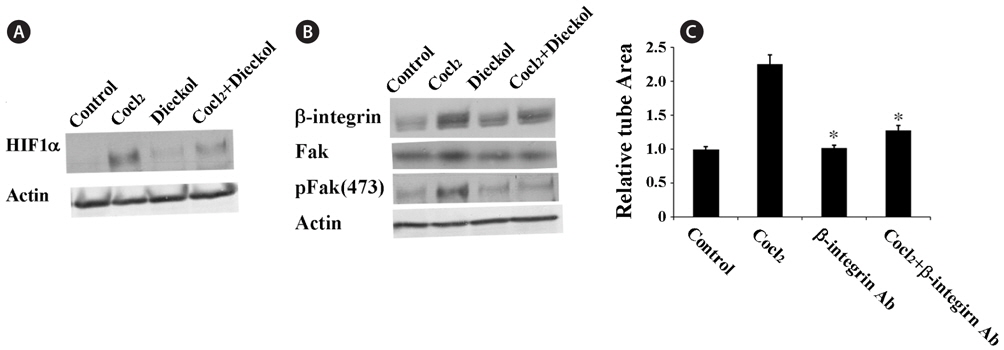
To understand the molecular mechanisms by which dieckol inhibits CoCl2-induced angiogenesis, we investigated the effect of dieckol on the expression of hypoxia-inducible factor 1-alpha (HIF1α) (Fig. 3A). HUVECs were treated with 100 μM CoCl2 in the presence or absence of 25 μM dieckol. CoCl2 treatment increased HIF1α expression approximately 2.8-fold compared with the PBS-treated control cells, and this effect was attenuated by dieckol. To further examine the molecular mechanisms by which dieckol inhibits angiogenesis in HUVECs, we investigated the effects of CoCl2 and dieckol on the expression of β1 integrin (Fig. 3B). β1 Integrin has been found to be responsible for cell proliferation, adhesion, and invasive migration during angiogenesis in response to hypoxic conditions (Niewiarowska et al., 2011; Li et al., 2012). The role of β1-integrin in CoCl2-induced hypoxic angiogenesis in HUVECs was also demonstrated in this study (Fig. 3C). HUVECs were pre-incubated with a specific antibody for β1-integrin before being treated with CoCl2. After treatment with the β1-integrin antibody, CoCl2 treatment failed to stimulate tube formation in HUVECs, indicating that β1-integrin plays a major role in CoCl2-induced angiogenesis. Based on this result, we tested the effects of CoCl2 and dieckol on the expression of β1-integrin in endothelial cells. As shown in Fig. 3B, CoCl2 effectively increased the expression of β1-integrin in HUVECs, and dieckol suppressed this effect. Additionally, CoCl2 treatment increased the expression and phosphorylation of focal adhesion kinase (Fak), a major downstream molecule of β1-integrin, and this effect was significantly inhibited by dieckol.
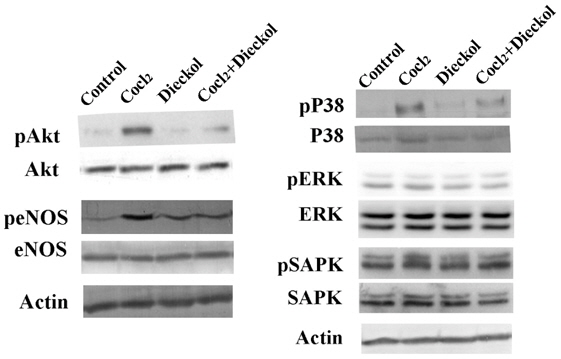
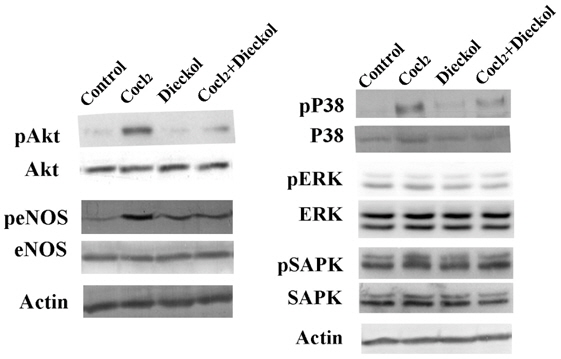
In addition, we determined whether dieckol regulates cellular signaling pathways involved in the response to hypoxic stress. The activation of Akt/NO signaling was measured, as well as the activation of mitogen-activated protein kinase (MAPK) family proteins p38 MAPK, extracellular regulated kinase (ERK), and stress-activated protein kinase (SAPK) (Fig. 4). CoCl2 treatment induced phosphorylation of Akt (Ser-473) and endothelial NO synthase (eNOS), and this phosphorylation was blocked by dieckol. CoCl2-induced p38 MAPK activation, but not CoCl2-induced ERK and SAPK activation, was inhibited by dieckol. Taken together, these data suggest that dieckol inhibits CoCl2-induced signaling downstream of HIF1α during hypoxia-induced angiogenesis in HUVECs via effects on β1-integrin, Fak, Akt/eNOS, and p38 MAPK.
Angiogenesis is essential for tumor growth and metastasis (Fidler and Ellis, 1994; Hanahan, 1997; Risau, 1997), and the development of new therapeutic drugs targeting angiogenesis has become a major focus of cancer research. Although accumulating evidence indicates that hypoxia-induced ROS signaling is associated with carcinogenesis and tumor angiogenesis (Liu et al., 2006; Zhou et al., 2007; Kim et al., 2006; Lo and Cruz, 1995), the precise mechanisms through which tumor formation and angiogenesis are regulated by endogenous ROS remain to be elucidated.
The primary regulator of hypoxia-induced angiogenesis is HIF1α (Jiang et al., 2001; Maxwell et al., 1997; Ferrara, 2005; Coultas et al., 2005). Under hypoxic conditions, ROS production facilitates HIF1α expression, which stimulates the expression of vascular endothelial growth factor (VEGF), which mediates tumor growth and angiogenesis (Medici and Olsen, 2012). ROS inhibitors decrease HIF1α expression and attenuate VEGF transcriptional activation (Xia et al., 2007), suggesting that endogenous ROS regulates levels of VEGF through regulation of HIF1α. In addition, several recent studies have reported that the cooperative activation of HIF1α, PI3K/Akt, and mTOR is required for the expression of fibronectin and β1 intergrin under hypoxic conditions (Distler et al., 2007; Lee et al., 2010). Hypoxia facilitates cellular adhesion to fibronectin and increases expression of β1-integrin, which mediates changes in cytoskeletal structure and cell migration under hypoxic conditions by activating Fak (Berry et al., 2000; Pirone et al., 2006). These reports are consistent with the results presented here, in which CoCl2-induced hypoxia increased cell adhesion and migration and expression of β1-integrin and Fak, whereas treatment of the cells with the antioxidant dieckol decreased hypoxia-induced cell adhesion, migration, and tube formation, as well as the expression of both β1-integrin and Fak. Tube formation and vessel sprouting induced by CoCl2 are also blocked by treatment with dieckol or an anti-β1-integrin antibody. These results suggest that CoCl2-induced hypoxia increases angiogenesis in HUVECs via effects on signaling through β1-integrin and Fak downstream of HIF1α. The Akt/NO pathway and mitogen-activated protein kinase (MAPK) signaling are also important mediators of hypoxia-induced angiogenesis. Inhibition of eNOS inhibits hypoxia-induced endothelial cell proliferation, migration, and tube formation in vitro, as well as angiogenesis in vivo (Papapetropoulos et al., 1997). Hypoxia activates eNOS and increases NO production in an Akt-dependent manner (Papapetropoulos et al., 1997; Zachary and Gliki, 2001; Min et al., 2004), and stimulates ROS generation in a MAPK-dependent manner (Millar et al., 2007), and these effects result in endothelial angiogenesis. Our results showed that treatment with the antioxidant dieckol decreased the hypoxia-induced activation of Akt and p38 MAPK in HUVECs. Taken together, our results suggest that dieckol inhibits signaling downstream of HIF1α, including signaling mediated by β1-integrin, Fak, Akt/eNOS, and p38 MAPK, and these effects of dieckol were associated with inhibition of CoCl2-induced angiogenesis in HUVECs. Thus, dieckol inhibits hypoxia-induced signaling in HUVECs through several defined molecular mechanisms. These results extend our understanding of the molecular mechanisms through which dieckol exerts its anti-tumor angiogenic effects, and suggest that dieckol is a potent HIF1α inhibitor and a potential chemotherapeutic agent in the clinic.