Use of universal primer targeted conserved regions in small subunit ribosomal RNA (SSU rRNA) gene has been utilized as a useful tool for identifying species and amplifying unknown functional gene (Pineau et al., 2004) as well. In 1989, Bottger (1989) first demonstrated that a portion of the 16S rRNA gene from Legjonella pneumophila,
PNA is an oligonucleotide analogue with polyamide backbone. Because of strong binding with DNA, this mimic DNA has been exploited to produce powerful bimolecular tools, antisense and antigene agents, molecular probes and biosensors (Nielsen and Egholm, 1999). Since PNA cannot extended by the polymerase, PNA has been also successfully utilized for clamping PCR amplification in mutation detection (Orum, 2000), and parasite detection (Troedsson et al., 2008b).
Recently, Vestheim and Jarman (2008) has proposed that a 3’-modified oligonucleotide, C3 spacer, can also be a useful clamping probe for PCR amplification of dominant gene. Inserted propyl group cannot be recognized by polymerase and thus isn’t accessible for further reactions. C3 is a cost-effective nucleotide. Comparing to PNA, C3 is much cheaper and relatively easy to synthesize. If C3 has a competitive clamping efficiency for dominant gene to PNA, it is favorable to use as a clamping probe for DHPLC assay.
Therefore, two different clamping probes were tested with same sequence, C3 spacer and PNA, in terms of clamping efficiency of dominant gene and enhancement of PCR amplification of rare genes using two distinct model assays, blue crab and grass shrimp.
A blue crab model (CS model),
The clamping probe was designed to prime onto antisense strand of the target gene and to prevent elongation of forward PCR primer. To avoid the any false priming to its parasitic gene fragment and primer dimer with PCR primers, candidate of probe sequence were analyzed by Primer premiere 5.0 (Premier Biosoft International, Palo Alto, CA, USA). C3 spacer was synthesized from Integrated DNA Technologies (Coralville, IA, USA): BC-1361F-C3, 5´- GGT GTC CAG TTC GCA G/C3SP/-3´ for blue crab,
To investigate the variation of clamping efficiency depending on clamping probe concentration, clamping-PCR was performed, with varying concentration (0 - 2 μM) of each probes, by a set of universal primer: Univ18s-1131F (5´-AAA CTY AAAGRA ATT GAC CC-3´) (Troedsson et al., 2008a) and Univ18s-1629R (5´-GAC GGG CGG TGT GTR C-3´) (Gruebl et al., 2002). The twenty μl of PCR reaction mix was constituted by 1X DiscoveraseTM PCR buffer (Invitrogen), 0.3U of DiscoveraseTM DNA Polymerase (Invitrogen), 0.5 μM of each Primer, varying concentration of clamping probe, 200 μM of dNTP, 2 mM of MgSO4 and 2 μl of templates. Thermal cycling condition was set as: 30 s at 94˚C; 25 cycles of 30s at 94˚C, 30s at clamping probe’s annealing temperature, 30s at 54˚C, 30s at 68˚C; and finally 5min at 68˚C. The annealing temperature for clamping probe was set to 66˚C for PNA and 48˚C for C3 in blue crab, 65˚C and 46.4˚C for grass shrimp, respectively. All clamping-PCR were carried out in these conditions. After clamping-PCR, PCR products were applied to DHPLC to quantify the amplicon concentration.
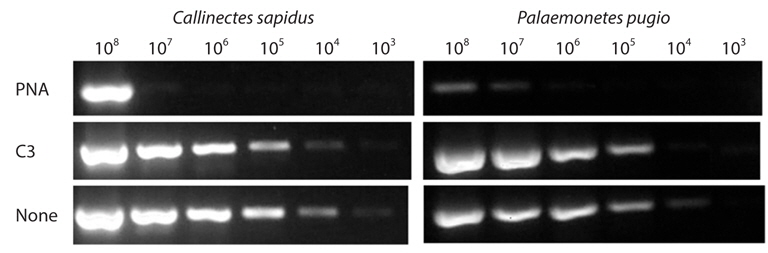
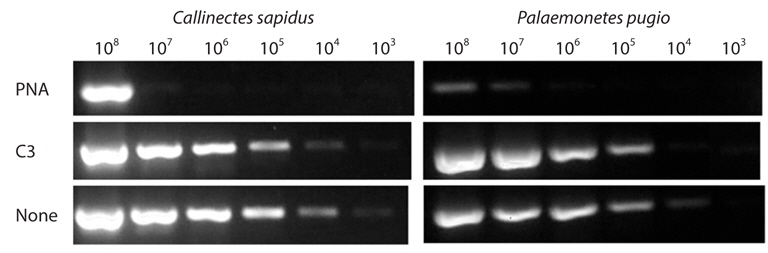
Clamping efficiency of each type of probe was investigated using serially diluted plasmid DNAs as templates (108 -103 copies). For this, each clamping-PCR were performed with previously described mixture adding 1 μM of clamping probe. The amplicons were eluted on 1.5% agarose gel.
>
Detection of rare gene by DHPLC
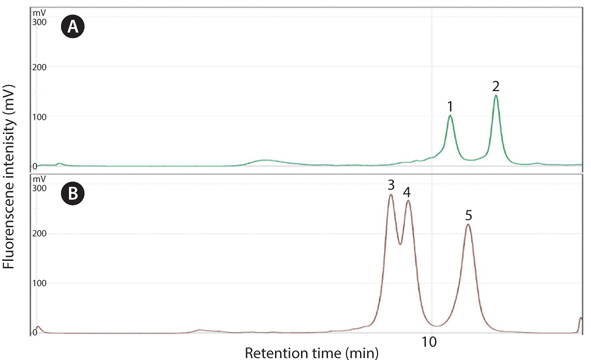
To investigate the competency of rare gene detection, clamping-PCR was carried out to amplify the rare gene with clamping the amplification of dominant gene with clamping probe. A 3 by 7 factorial matrix mixture was used as a template. For detection of rare gene, PCR amplicon was further separated by DNASep HT Cartridge (Transgenomic, Omaha, NE) with linear gradient elution of WAVE® Optimized Buffers. Separation condition was tested by varying oven temperature and buffer B% (Troedsson et al., 2008a; Cho et al., Unpublished) and for 10 min. Chromatographic analysis was carried out by HSX-3500 fluorescence detector (Ex = 495 nm and Em = 537 nm) after SYBR Gold staining (Molecular Probe Inc.) with Navigator software version 1.6.2 (build 12) (Transgenomic, Omaha, NE).
All data given in mean value and standard deviation from, at least, duplicate data. The significance of clamping efficiency depending on probe concentration was analyzed by analysis of variance (ANOVA) and tukey’s test for post-hoc test for mean difference. Nonlinear regression analysis was also carried out for amount of target-amplicon from varying concentration of clamping probe. All statistical analyses were performed by statistical software, Sigmastat 3.11 (Systat Software, Inc.).
To quantitatively estimate the clamping efficiency for each type of clamping probe, serially diluted plasmid DNAs were applied to clamping PCR with/without 1 μM of clamping probe. In CS model, PNA completely blocked amplification of target amplicon up to 106 copies of template, which were 5 orders of magnitude than PCR amplification without clamping probe. In other hand, C3 rarely showed clamping efficiency. In PP model, up to 105 copies, PNA completely blocked and even slightly in 108 and 107 copies, band intensity of target amplicon was significantly smaller than those of none clamping probe PCR condition and C3 amplification (Fig. 1). The clamping efficiency of PNA was always superior to C3 spacer by more than 3 orders magnitude.
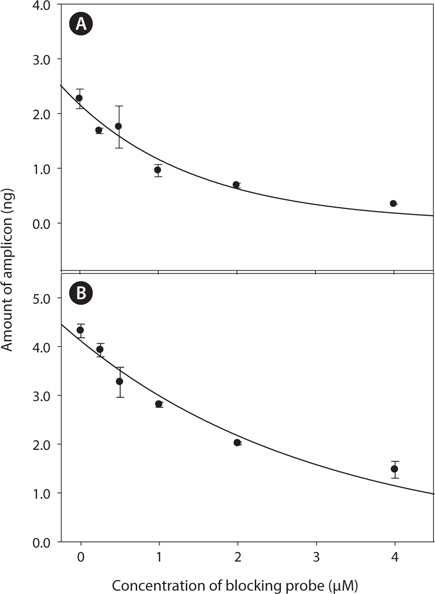
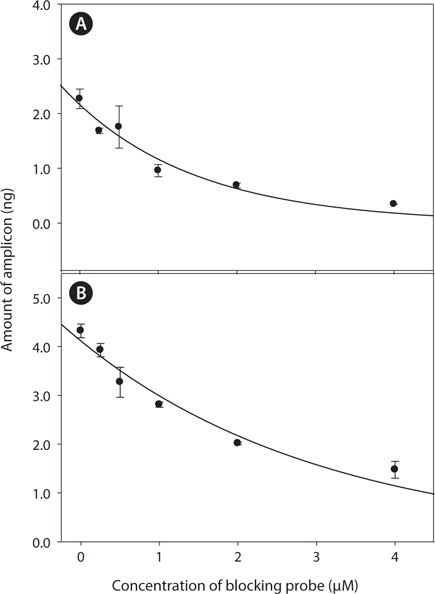
Optimal concentration for clamping probe was tested by varying concentration of clamping probes from 0 μM to 2 μM. PCR amplicons were further injected to DHPLC to quantify the amount of target amplicons in response to probe concentration. With increase of clamping probe, amplification of target-amplicon was obviously suppressed in both two model system (
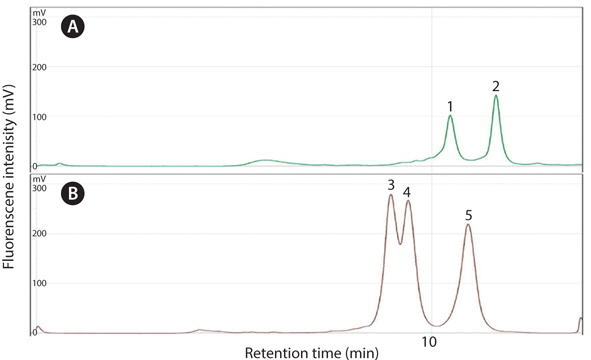
Optimization of dHPLC assay was carried out with varying buffer B% and oven temperature conditions. For CS model, separation was optimized at 59.5˚C of oven temperature and 60-70% of linear gradient buffer B at 3.5 mL/min of flow rate (Troedsson et al., 2008a) while, for PP model, 57.5˚C and 62-68% at 0.45 mL/min (Cho, 2012). Retention time of each host and parasite peaks were highly reproducible (Fig. 3). WE applied these assay for further separation of PCR amplicons from pDNA mixture to measure the enhancement of rare gene amplification.
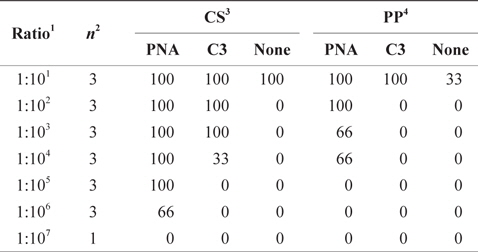
Detection of rare gene was highly improved by clamping- PCR, especially by PNA- clamping PCR. In CS model, the parasite,
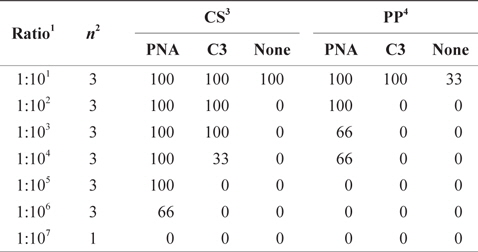
Detection of rare gene (%) with clamping probes in multi template plasmid DNA mixtures by PNA-PCR, C3-PCR and PCR coupled with DHPLC assay
In aquatic ecosystem, animals are always exposed to relatively large biomass of parasitesand co-existent with diverse symbionts in various ways (Kuris et al., 2008). Except epidemic case, detection of unknown symbionts is highly time- and cost-consuming work because of their rare intensities. Recently, denaturing high-performance liquid chromatography (DHPLC) has been utilized as a new diagnostic technique for detecting parasitism in marine eukaryotic organism (Troedsson et al., 2008a; Troedsson et al., 2008b). The potential of this new technology is now expanding its application for marine biology and ecology. PCR-based DHPLC assay has been turned out a versatile and efficient model for detecting low ranged parasites and , with universal primers targeted highly conserved region, successfully utilized to investigate the symbiont composition in 18s rRNA for eukaryotes (Schmidt and Relman, 1994) and 16s rRNA for bacteria (Amann and Ludwig, 2000). To achieve the amplification of rare parasitic gene, however, it is necessary to suppress the amplification of the dominant host gene to avoid a biased PCR amplification of the dominant host gene.
C3 (Dames et al., 2007; Vestheim and Jarman, 2008) and PNA (Orum, 2000; Shakeel et al., 2006; Troedsson et al., 2008b) have been used as effective clamping probes and further enhancer of PCR amplification of rare gene. The clamping efficiency of PNA was obviously superior to C3 spacer by 2-3 orders of magnitude (Fig. 1). PNA has a strong binding independent of salt concentration and also greater specificity in binding to complementary DNA (Demidov and Frank-Kamenetskii, 2004). This high specificity and strong binding may facilitate strong hybridization on intra-strand of target DNA and prevent polymerase to displace the PNA molecules from the template (Shakeel et al., 2006).
In this study, the extension temperature (68˚C) is much higher than annealing temperature of any C3 probes. During extension, the polymerase may displace C3 probe from the template, and then extend towards completion of the reaction (Shakeel et al., 2006). When the binding position of clamping probe is closer to PCR primer, the DNA/DNA binding activity may be relatively stronger than further one before thermal cycling temperature stabilized.
PNA binding should precede PCR primer binding. Tm of the PNA and DNA is, therefore, the most important variables that affect binding order, which should be between 65˚C and 75˚C (Orum, 2000). Mostly, PNA can meet this condition with relatively short sequence because of its stronger PNA/DNA binding. As a general rule, A Tm of a PNA/DNA duplex is 1˚C higher per base pair than Tm of the corresponding DNA/DNA duplex (in 100 mmol dm-3 NaCl) (Shakeel et al., 2006). To make C3 spacers of PNA’s Tm, the sequence should be prolonged much more than PNA length. Considering interaction with PCR primers and various symbiont and host genes, long clamping probe may possibly generate non-specific annealing and a dimer with PCR primers, which eventually decrease the detection efficiency of clamping-PCR. On the order hand, PNA can reach the Tm by only 12-18 bases with much stronger binding activity. Therefore, PNA can be a more efficient clamping probe for enhancement of PCR amplification of rare gene than C3 spacer.
The ratio of clamping probe to template is constantly varied with each cycles and kinetic of PNA annealing are constantly changing in the reaction (Demers et al., 1995). Therefore, the optimal concentration of clamping probe should be determined carefully and fully tested with various conditions. In the study, amount of target PCR amplicon was exponentially decreased with increase of clamping probe concentration (Fig. 2) in accordance with previous study (Demers et al., 1995). One μM of PNA concentration has been successfully utilized for parasitic gene detection in wild blue crab samples ((Troedsson et al., 2008a; Troedsson et al., 2008b) and showed over 50% inhibition efficiency in target gene amplification in the study. Therefore, this concentration was applied to further experiment for clamping efficiency.
A significant difference was observed at clamping efficiency between two PNA probes. In CS model, BC18S-1361-PNA completely blocked amplification of pDNA of
Estuarine and coastal environments can be good habitats and nursery ground for many aquatic biotas and also be a sink tank for land-driven compounds. Monitoring of these contaminants dynamics is crucial not only for concern of human health but also conservation of ecosystem homeostasis. Input of contaminants may make hosts more susceptible to parasitism and increase mortality rates of infected hosts in the ecosystem (Möller, 1987; Lafferty and Kuris, 1999), although being disagreements also available on this matter (Lafferty and Kuris, 1999). Tremendous effort has, so far, been made to understand the interaction between environment contamination and parasitic epidemics in aquatic ecosystem (Lafferty and Kuris, 1999; Kim et al., 2008). Detection or discovery of parasitism is probably a crucial prerequisite condition to understand the impact of environmental stress on aquatic ecosystem. During the past decades, noticeably developed diagnostic techniques have enabled detection and quantification of a variety of parasites, parasitoids and pathogens including molecular diagnosis (Lightner and Redman, 1998; Cunningham, 2002; Mothershed and Whitney, 2006; Thakur et al., 2008). DHPLC is also one of those versatile and competent tools for monitoring parasitic/symbiont composition. In order to facilitate the detection of rare symbiont gene, clamping- PCR is a promised tool to overcome the biased amplification of PCR amplification. Therefore, PNA can be the most useful and prominent tool for clamping-PCR over its high price and complexity of synthesis.