Along with that multidimensional separation platforms coupled with advanced mass spectrometry (MS) have been continuously developed for enlarging the separation capacity, that provides the enhancement of proteomic datasets in the identification of proteins out of biological specimens (
From these aims mentioned above, a number of stable isotope labeling methods are introduced and verified for obtaining more reliable quantification of proteome samples with high precision and accuracy, compared to label-free experiments.4 Differed from
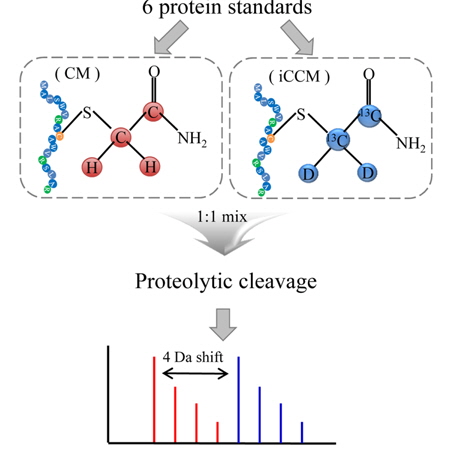
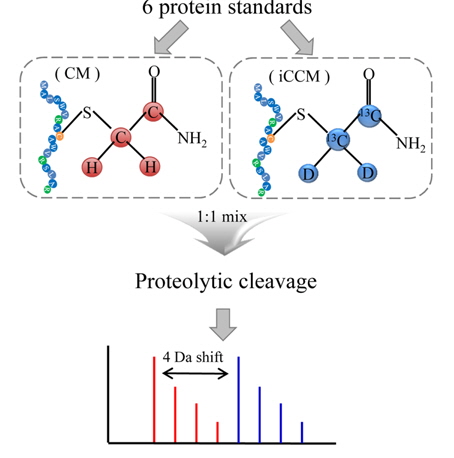
In this study, we presented a new isotope-coded carbamidomethylation (iCCM)-based isotope labeling for the quantitative proteomics with inexpensiveness, ease of use, simplicity, and robustness. In iCCM-based isotope labeling, the two protein mixtures originated from two different states, for instance, healthy versus cancer sera, are individually isotopically labeled with iodoacetamide (IAA) and its isotope (IAA-13C2, D2), denote as CM and iCCM, respectively, by means of covalently reacting with the cysteinyl residues in proteins, leading to a mass shift of all cysteinyl residues to be + 4 Da. Finally, two protein samples are mixed equally, proteolytically digested, and then quantified via a comparison of the relative abundance ratios of the corresponding pair ions or their fragment ions obtained by nLC-ESI-MS/MS run. To evaluate the efficiency of iCCM-based labeling with respect to reproducibility and precision in protein quantification, two aliquots (43 mg) of 6 protein standards, composed of bovine serum albumin (BSA, 64.4 kDa), serotransferrin (77.1 kDa), lysozyme C (16.2 kDa), beta-lactoglobulin (19.2 kDa), beta-galctosidase (116.5 kDa), alpha-lactalbumin (16.2 kDa), were individually treated with IAA and its isotope, respectively. Each of CM- and iCCM-labeled protein standards was pooled equally, proteolytically cleavaged, and followed by which the resulting peptides were analyzed using a nLC-ESI-MS/MS run in order to evaluate the efficiency of iCCM-based relative quantification, compared to that of mTRAQ using the same protein standards. Finally, we applied the developed iCCM-based quantitative method to human lung cancer serum proteomics.
Ammonium bicarbonate (ABC), dithiothreitol (DTT), formic acid, iodoacetamide (IAA), iodoacetamide-13C2, d2 (isotope labeled-IAA) and L-cysteine and dimethyl sulfoxide (DMSO) were purchased from Sigma (St. Louis, MO, USA). Sequencing grade trypsin and Peptide Nglycosidase F (PNGase F) were obtained from Promega Corp. (Madison, WI, USA). Oasis HLB cartridge was purchased from Waters (Milford, MA, USA). The mTRAQ kit and the 6 protein standards delivered with the iTRAQ kit were provided by AB Sciex (Framingham, MA, USA). HPLC-grade acetonitrile and water for a binary gradient elution were obtained from Burdick & Jackson (Ulsan, Korea). Fused-silica capillaries (25, 50, 75, and 100 mm-i.d.; 365 μm-o.d.) used for the capillary LC column and tubing connections were obtained from Polymicro Technology LLC (Phoenix, AZ, USA). Magic C18AQ (3 μm, 100 Å) and Magic C18AQ (3 μm, 200 Å) resins were delivered from Michrom BioResources Inc. (Auburn, CA, USA). Fittings, adapters, and PEEK tubing were purchased from Upchurch Scientific® (Oak Harbor, WA, USA) of IDEX Health & Science LCC.
>
Tryptic digestion for iCCM-based quantification
Prior to tryptic digestion, two aliquots (43 mg) of the 6 protein standards or human plasma sample was denatured and reduced with 50 mM ABC contained with 10 mM DTT for 2 hours at 37℃. In order to alkylate the remaining thiol group, two aliquots were individually treated in an ice bath for 2 hours in the dark with 20 mM IAA and isotope labeled-IAA, respectively. L-cysteine (40-fold access of IAA) was added to both aliquots to remove excess IAA and its isotope at room temperature for 30 min. The resulting peptide digests labeled individually with CM and iCCM were mixed at the ratio of 1:1, and then digested with sequencing graded modified trypsin at a ratio of 50 : 1 (protein : trypsin, w/w) at 37℃ for 18 hours. In order to stop the tryptic digestion, the solution was treated with 1% formic acid (v/v). The digested peptides were stored in -80℃ before use. For human sera samples, the endoglycosidic treatment, in particular, was first carried out using a PNGase F, prior to tryptic proteolysis, to minimize the occurrence of trypsin miscleavage that might be caused by a steric hindrance of N-linked glycans covalently attached to asparagine (N) with the consensus motif as N-X-S/T (X for any amino acid except proline).
Prior to mTRAQ-based stable isotope labeling, the same 6 protein standards as used in iCCM-based isotope labeling was tryptically digested and desalted by an Oasis HLB cartridge to remove chemical hindrances (i.e., thiols or primary amines) containing in protein solution. Eluted peptides were lyophilized using SpeedVac, and reconstituted with 50 mM triethyl ammonium bicarbonate. Two aliquots (43 μg) of tryptic digests from 6 protein standards were isotopically labeled with each of mTRAQ reagents (Δ0 and Δ4) with a 50 uL isopropanol for 1 hour at room temperature. Each of light (Δ0)- and heavy (Δ4)-labeled digests was re-lyophilized, reconstituted with 0.1% formic acid solution, and then pooled equally for next shotgun proteomic analysis.
nLC-ESI-MS-MS was carried out by a binary gradient elution and the model 1260 capillary LC system from Agilent Technologies coupled to a Q-Exactive Fourier Transform orbitrap-mass spectrometer (FT orbitrap-MS) from Thermo Scientific (Bremen, German) via electrospray ionization. The pre-column (RP) to trap sample was used with an analytical capillary RPLC column (150 mm × 75 μm-i.d.). A pulled tip analytical capillary column used in this study was prepared by packing with Magic C18AQ (3 μm, 100 Å) resins. For the purpose of RPLC separation for peptides mixture, the trap column was prepared in a capillary (100 μm-i.d., 360 μm-o.d.) with an end frit (2 mm in length), and the capillary was packed in sequence with Magic C18AQ (3 μm, 200 Å) resins for 0.5 cm. The trap column and the analytical column were connected via a PEEK microcross.10
For the RPLC runs, two buffer solutions, (A) 2% ACN in water and (B) 93% ACN with 5% DMSO in water, both with 0.1% formic acid, were used as mobile phase solutions. The flow rate for RPLC run was 200 nL/min and the binary gradient run conditions were as follows: Samples were loaded into the trap column first by 0% B for 10 min, and initial increase to 8% B for 1 min then linearly increased to 15% up to 20 min., to 32% up to 55 min, ramped to 80% for 3 min, was held at 80% for 10 min to clean the RP column, and finally it was decreased to 0% over 2 min, and was held for 20 min to re-equilibrate the RP column. In human lung cancer proteomic, the RPLC runs were used the same as mentioned above, excepting that the gradient conditions was modified to be 25% B up to 115 min. from 32% B up to 55 min. The eluted peptides from the capillary column were directly fed into the FT orbitrap-MS via electrospray ionization in positive ion mode with 270℃ of capillary temperature and a voltage of 2.5 kV was applied through the Pt wire connected to the microcross. The column outflow rate was 200 nL/min by spliting the capillary (25 μm-i.d) attached to the microcross for MS precursor scan (
The developed iCCM-based isotope labeling strategy presented in this study was aimed for providing more reliable quantitative datasets in proteomic analysis with high accuracy and precision, compared to conventional nonisobaric isotope labeling approaches. In particular, it is characteristic that an iCCM-based isotope labeling of proteome was easily achieved during general proteolysis without any other additional purification steps, as shown in Figure 1, whereas mTRAQ-based approach might be inevitably accompanied with buffer exchange step to remove several interfering substances (
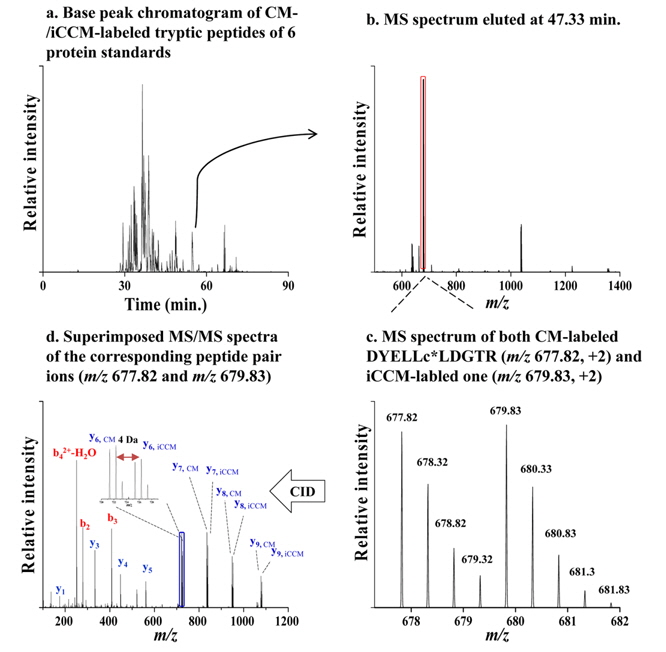
Figure 2 shows a base peak chromatogram (BPC) obtained from nLC-ESI-MS/MS experiments of the tryptic peptides (300 ng) derived from iCCM-based isotope labeling (Figure 2a), along with the MS scan spectrum eluted at tr = 47.33 min (Figure 2b). Out of the corresponding peptide pair ions detected in Figure 2b, both of
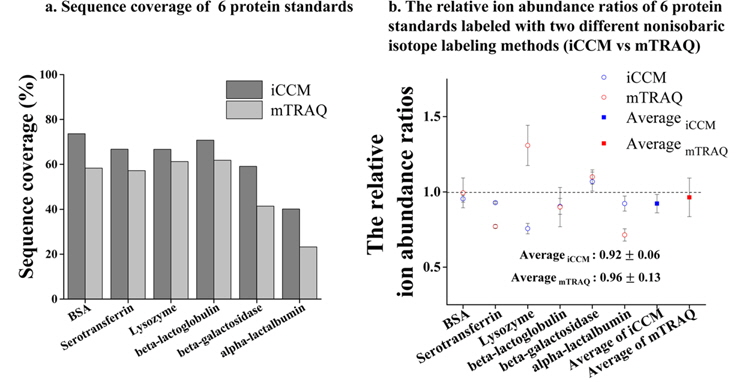
For the purpose of further evaluation, the same protein standards as used in former experiment were isotopically labeled using mTRAQ to make a comparison between the developed iCCM-based quantification and that of mTRAQ with respect to the efficiencies in a qualitative/quantitative experiment. Figure 3 shows the peptide sequence coverage of 6 protein standards and their relative abundance ratios of identified corresponding peptide pair ions differentially isotope-labeled with each of iCCM and mTRAQ. Based on the our quantitative experiments in triplicate, it is shown that the performance of an iCCM-based isotope labeling approach is more superior to that of mTRAQ in both of a number of identified peptides and the reproducibility in quantitative variations. Resultingly, it is impressive that each sequence coverage of 6 protein standards was averagely more improved to at least ~10% than that of mTRAQ (Figure 3a). Furthermore, the standard deviation (S.D.) of 0.06 in iCCM reduced nearly two-fold less than that (0.13) of mTRAQ on the basis of the observed ratios of the corresponding peptide pair ions from 6 protein standards (Figure 3b), while it shown that each of the median observed ratios between those iCCM and mTRAQ was similar to be 0.92 for iCCM and 0.96 for mTRAQ, respectively. For the reason of which mTRAQ is a bit as lacking in quantitative reproducibility, it might be caused by two major reasons; i) the interfering substances (
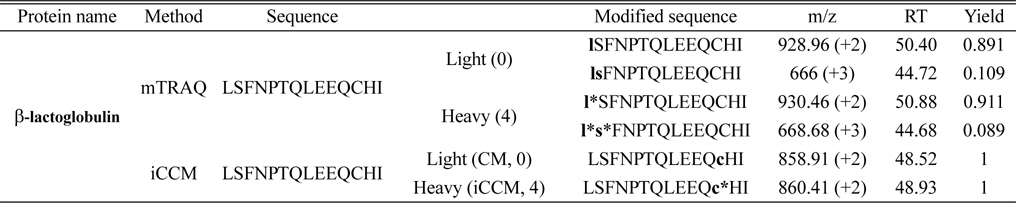
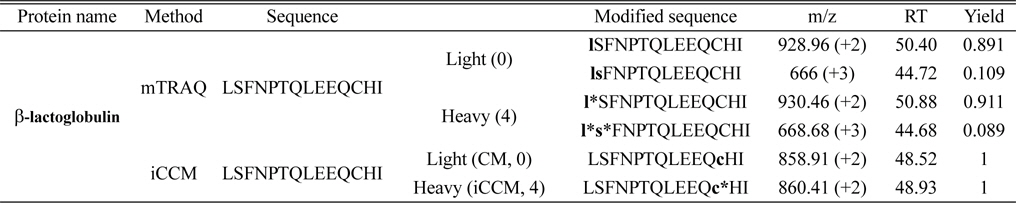
Comparison of three tryptic peptides of beat-lactoglobulin labeled with mTRAQ and iCCM and their modified sequences assigned using Proteome Discoverer software (version 1.3.0.339)
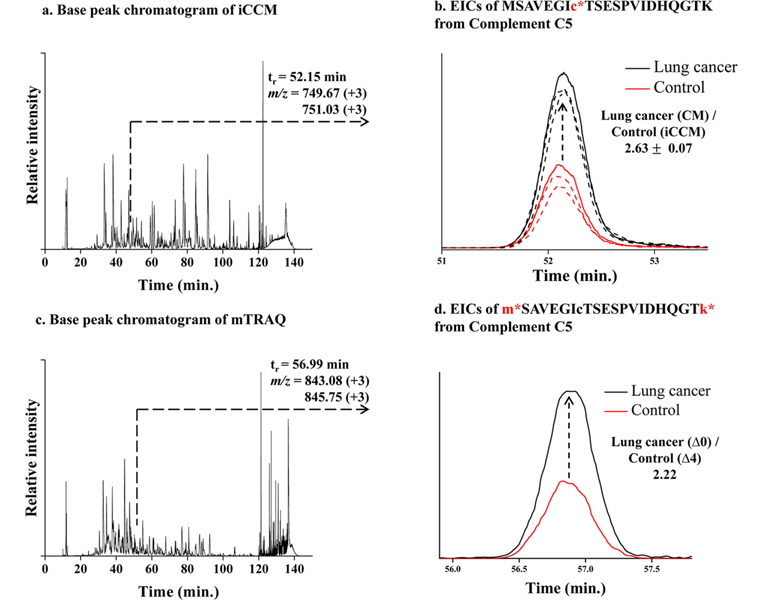
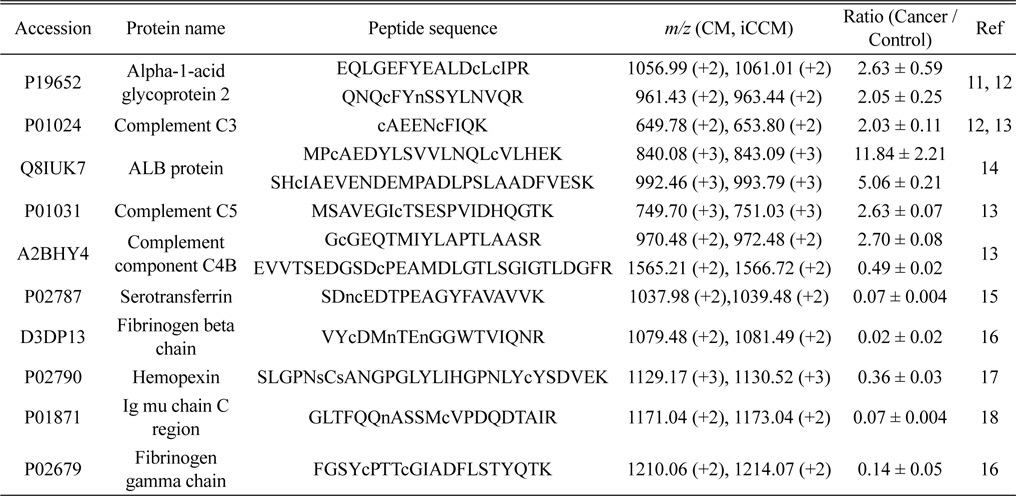
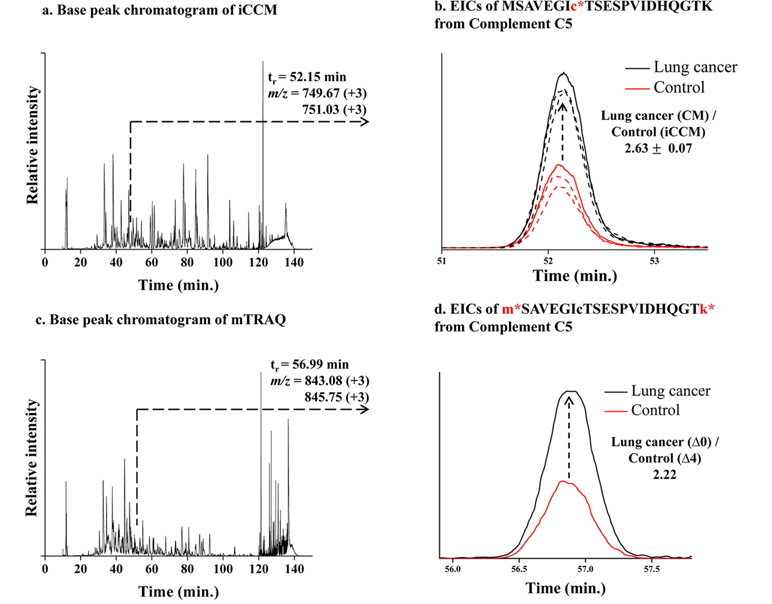
Finally, we applied the developed iCCM-based stable isotope labeling to lung cancer serum proteome in order to assess the usefulness in the quantitative profiling of a large scaled protein samples. We first prepared the CM-/iCCM-labeled protein mixtures by which the liver cancer sera and controls are isotopically labeled separately with CM and iCCM, respectively, mixed with the ratio of 1:1 (lung cancer:control), and then subjected to the proteolytic treatment with a trypsin. Finally, the resulting CM-/iCCMlabeled digests were introduced to nLC-ESI-MS/MS experiments. Furthermore, the same sera samples as used in iCCM labeling also prepared via a non-isobaric labeling with mTRAQ in order to compare with quantitative results from those iCCM. The two BPCs obtained by shotgun proteomic analyses of each of iCCM-labeled peptides and mTRAQ-labeled one, as shown in Figure 4a and 4c, respectively. From shotgun analyses, 158 proteins from 378 peptides, including at least one or more cysteine residues, were identified out of total 243 proteins and simultaneously allowed the quantitative profiling of up-/down-regulated peptides through measuring the observed ratios of CM-/iCCM-labeled corresponding pair ions. As a result, we found that 13 peptides from 10 proteins were changed more than 2- folds in the lung cancer proteome sample compared to the control and they are listed in Table 2. Among them, both ions of
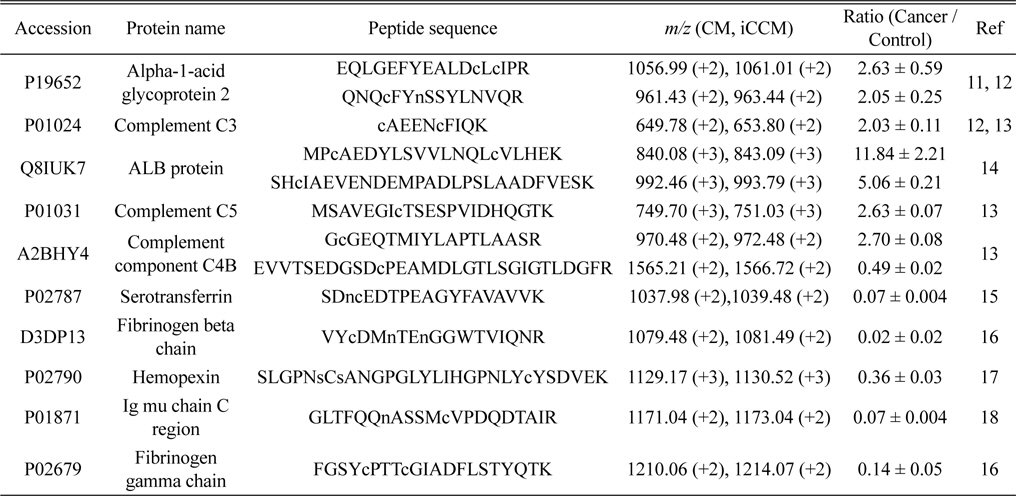
A total 13 peptides (from 10 proteins) showing more than 2-folds of change in their levels of lung cancer patient samples compared to the control obtained by measuring the relative abundance ratios of CM-/iCCM-labeled peptide pair ions
In this study, we introduced a new iCCM-based isotope labeling strategy can be considered as one of promising strategies for quantitative proteomics, compared to conventional isotope labeling methods. Several advantages of iCCM-labeling method can be summarized as follows: i) stable isotope labeling in protein level can bypass the chances of unequal digestion in two different protein samples or of unequal retrieval of peptides compared to those stable isotope labeling strategies in peptide level, ii) there is not necessary of any purification step to remove some chemical hindrances in conventional isotope labeling methods, and iii) light- and heavy-IAA reagents required for iCCM are much cheaper than typical isotope labeling reagents that can be easily accessible. First evaluation of iCCM-based stable isotope labeling was carried out by means of which the tryptic peptides mixed equally with both of CM- and iCCM-labeled protein 6 protein standards and then quantitative analyzed using nLC-ESI-FT orbitrap-MS/ MS. From our shotgun proteomic experiments, the peptide mixtures labeled with CM- and iCCM, having a mass difference of + 4 Da between them, can be simultaneously identified and quantified during MS/MS experiments. In addition, even though iCCM-based isotope labeling method presented in this study has the limitation of which only proteolytic peptides containing cysteinyl residues are meaningful in the quantitative examination, iCCM-based isotope labeling in protein level spontaneously allows to the minimization of quantitative variations caused by the irreproducibility of the activity of proteases and the recovery of vial-to-vial, compared to mTRAQ whereby the nonisobaric labeling is carried out in peptide level. Consequently, we confirmed that an iCCM-based isotope labeling can be considered as a complementary method for the quantitative proteomics.
![Base peak chromatogram (BPC) obtained from nLC-ESI-MS/MS run of the tryptic peptides (100 ng) derived from iCCM-based sample treatment (a) along with the MS scan spectrum eluted at tr = 47.33 min (b). MS spectrum of both m/z 677.82 [M+2H+]2+ and m/z 679.83 [M+2H+]2+ observed with the mass difference of 2.01 Da between both ions (c), and simultaneously identified via a MS/MS experiments as DYELLcLDGTR for CM-labeled peptide and DYELLc*LDGTR for iCCM-labeled one from serotransferrin, respectively (d).](http://oak.go.kr/repository/journal/14484/E1MPSV_2014_v5n3_63_f002.jpg)