To explore marine microorganisms with medical potential, we isolated and identified marine bacteria from floats, marine algae, animals, and sponges collected from Jeju Island, Korea. We isolated and identified 21 different strains from the marine samples by 16S rRNA analysis, cultured them in marine broth, and extracted them with ethyl acetate (EtOAc) to collect secondary metabolite fractions. Next, we evaluated their anti-oxidative and anti-inflammatory effects. Among the 21 strains, the secondary metabolite fraction of Bacillus badius had both strong antioxidant and anti-inflammatory activity, and thus was selected for further experiments. An antioxidant compound detected from the secondary metabolite fraction of B. badius was purified by preparative centrifugal partition chromatography (n-hexane:EtOAc:methanol:water, 4:6:4:6, v/v), and identified as diolmycin A2. Additionally, diolmycin A2 strongly inhibited nitric oxide production. Thus, we successfully identified a significant bioactive compound from B. badius among the bacterial strains collected from Jeju Island.
Marine organisms represent a promising future source of natural products due to the incredible diversity of chemical compounds they contain. Since the mid-1970s, marine environments have been systematically investigated as a source of novel biologically active agents, and marine bacteria, fungi, algae, sponges, coelenterates, seahares, bryozoans, tunicates, and nudibranchs have been especially studied (Mearns-Spragg et al., 1998; Schmitt et al., 2012).
Marine organisms have recently been shown to be a rich source of structurally unique and biologically active secondary metabolites. Among these, marine bacteria represent a major source of natural products with diverse chemical structures and biological activities (Kim et al., 2008), and are regularly found both on and within marine invertebrates. Marine and terrestrial bacteria differ due to the influence of their respective environmental conditions (ZoBell and Johnson, 1949). Microorganisms living in the sea must survive and grow in a marine environment with low nutrition, high salinity, and high pressure. To survive under such conditions, marine bacteria often produce compounds with unique biological properties, which are attractive to researchers. Marine
All solvents used for crude sample preparation were of analytical grade (Daejung Chemicals & Metals Co., Seoul, Korea). Lipopolysaccharide (LPS) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) penicillin streptomycin, and trypsin-EDTA were obtained from Gibco/BRL (Grand Island, NY, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5 -diphenyltetrazolium bromide (MTT), 1,1-diphenyl-2-picrylhydrazyl (DPPH), dimethyl sulfoxide (DMSO), and all other reagents and solvents were also purchased from Sigma Aldrich.
>
Collection and identification of marine bacteria
We collected four types of samples, from floats, marine algae, animals, and sponges from around the coast of Jeju Island, Korea in August 2011. Thereafter, all samples were rinsed three times with sterile seawater for 12 h at room temperature to remove loosely attached microorganisms. Samples were aseptically cut into small pieces using sterile surgical scissors and carefully placed onto prepared Petri-dishes containing Marine Agar 2216 (BD, Sparks, MD, USA) media. These plates were sealed and labeled by sample code and initial culturing date. Next, they were incubated at 29°C with humidity. After 3-5 days, cultivated bacteria were identified to the species level by PCR amplification of the 16S rRNA gene, BLAST analysis, and comparison with sequences in the GenBank nucleotide database. In this manner, we isolated 21 bacterial strains, and deposited voucher specimens in the Marine Bio-Resource Technology Laboratory of Jeju National University.
>
Preparation of secondary metabolite extracts
A single colony from a well grown agar plate was used as an inoculum and transferred to 300 mL Erlenmeyer flasks with 200 mL of Marine Broth 2216 (BD) medium containing 216peptone (0.5%), yeast extract (0.1%), and seawater (100%) for the production of secondary metabolites. These liquid culture flasks were incubated on a rotary shaker at 121 rpm and 29°C with humidity for 10 days. Thereafter, the broth was centrifuged (10,000 rpm, 15 min) to remove cells. Next, the supernatants were extracted with an equal volume of ethyl acetate (EtOAc; 200 mL). After separation, the organic phases were concentrated
>
Hydrogen peroxide (H2O2) scavenging activity
H2O2 scavenging activity was determined according to the method of Müller (1985), wherein 100 μL of 0.1 M phosphate buffer (pH 5.0) and the sample solution were mixed in a 96 microwell plate. Next, 20 μL of H2O2 was added, and the mixture, was incubated at 37°C for 5 min. Finally, 30 μL of 1.25 mM ABTS and 30 μL of peroxidase (1 unit/mL) were added, and the mixture was again incubated at 37°C for 10 min, after which we measured the absorbance with an enzyme-linked immunosorbent assay (ELISA) reader at 405 nm.
We purchased the murine macrophage cell line RAW 264.7 from the Korean Cell Line Bank (KCLB, Seoul, Korea). RAW 264.7 cells were cultured in DMEM supplemented with 100 U/mL of penicillin, 100 μg/mL of streptomycin and 10% FBS. Cells were incubated in an atmosphere of 5% CO2 at 37°C and subcultured every 2 days.
>
Determination of nitric oxide (NO) Production
Anti-inflammatory effects were evaluated following the protocol of Lee et al. (2013), with some modifications. RAW 264.7 cells (1 × 105) were plated and incubated with samples in the absence or presence of LPS (1 μg/mL) for 24 h at 37°C, and the supernatant of the cultured cells was used for NO quantification. A mixture of 100 μL of the supernatant and 100 μL of Griess reagent was incubated at room temperature for 10 min and the absorbance at 540 nm was measured on a microplate reader. We performed MTT and lactate dehydrogenase (LDH) assays to measure cytotoxicity: RAW 264.7 cells (1 × 105 cells/mL) plated in 24-well plates were preincubated and subsequently treated with LPS (1 μg/mL) coupled with samples at 37°C for 24 h. Next, 100 μL of MTT stock solution (2 mg/mL) was applied to the wells, and after 4 h of incubation, the supernatants were aspirated. The formazan crystal in each well was dissolved in 200 μL of DMSO and the absorbance measured via ELISA at a wavelength of 540 nm. The medium was carefully removed from each well, and LDH activity in the medium was determined using an LDH cytotoxicity detection kit.
>
Online high-performance liquid chromatography (HPLC)-ABTS+ assay
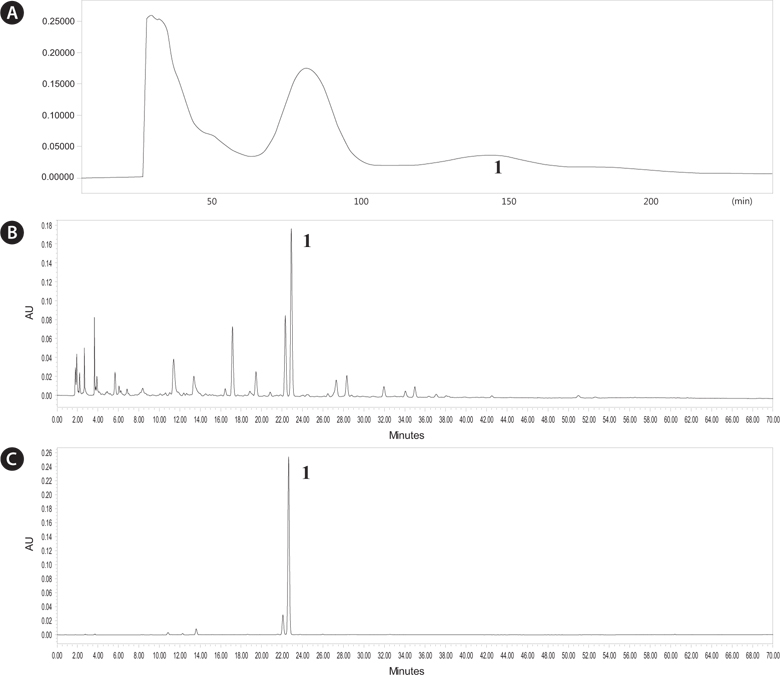
HPLC and ABTS assays were performed according to the method of Lee et al. (2013), with some modifications. For HPLC processing, the mobile phase comprised several ratios of acetonitrile to water in gradient mode as follows: acetonitrile: water (0-50 min: 20:80→60: 40, v/v; 50-60 min: 60:40, v/v; and 60-70 min: 100:0, v/v). The flow rate was 0.2 mL/min and the ultraviolet (UV) absorbance was detected at 280 nm.
>
Preparative centrifugal partition chromatography (CPC) separation procedure
The CPC experiment was performed using the assay of Lee et al. (2014), and the two-phase solvent system was composed of
All measurements were made in triplicate and all values are represented as means ± standard error (SE). The results were subjected to an analysis of variance (ANOVA) using Turkey’s HSD post hoc test to analyze the differences between groups, with a significance level of 0.05.
>
Culture and isolation of marine bacteria
The marine bacteria collected from the floats, marine algae, animals and sponges were cultured in marine agar media, and we successfully isolated 21 bacterial strains exhibiting morphological differences. We identified these bacterial strains to 17 genera: five belonged to the genus
[Table 1.] Scientific names of the 21 marine bacteria species isolated from marine samples
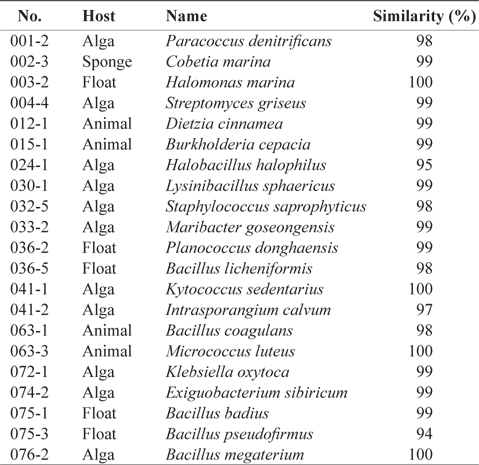
Scientific names of the 21 marine bacteria species isolated from marine samples
To evaluate the biological activity of the secondary metabolites extracted from the bacterial strains, all strains were cultured in flasks with 200 mL of liquid culture medium. After 10−15 days, the media and bacterial strains were extracted by adding EtOAc in preparation for further experiments.
>
Evaluation of biological effects of the extracts
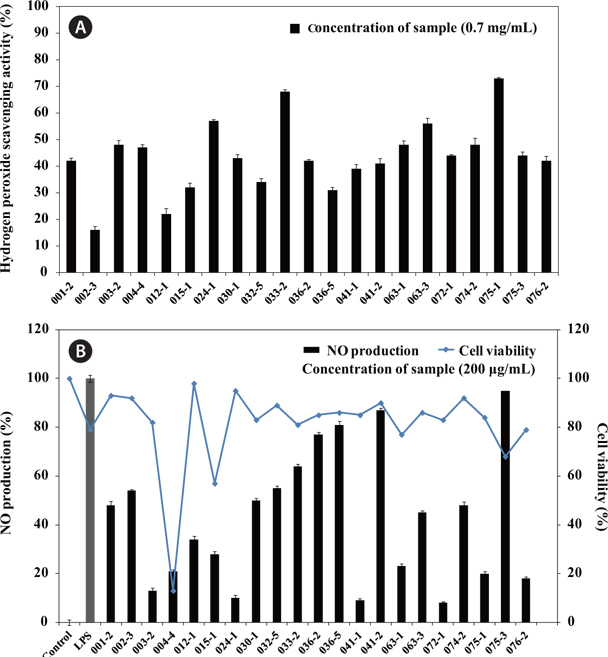
The results for H2O2 scavenging activity of the extracts containing secondary metabolites are shown in Fig. 1A. The secondary metabolite extracts from
>
Online HPLC-ABTS+ analysis of the of B. badius extract
We analyzed chromatography peaks and their radical scavenging activities in the
>
Purification and identification of anti-oxidative compounds from B. badius
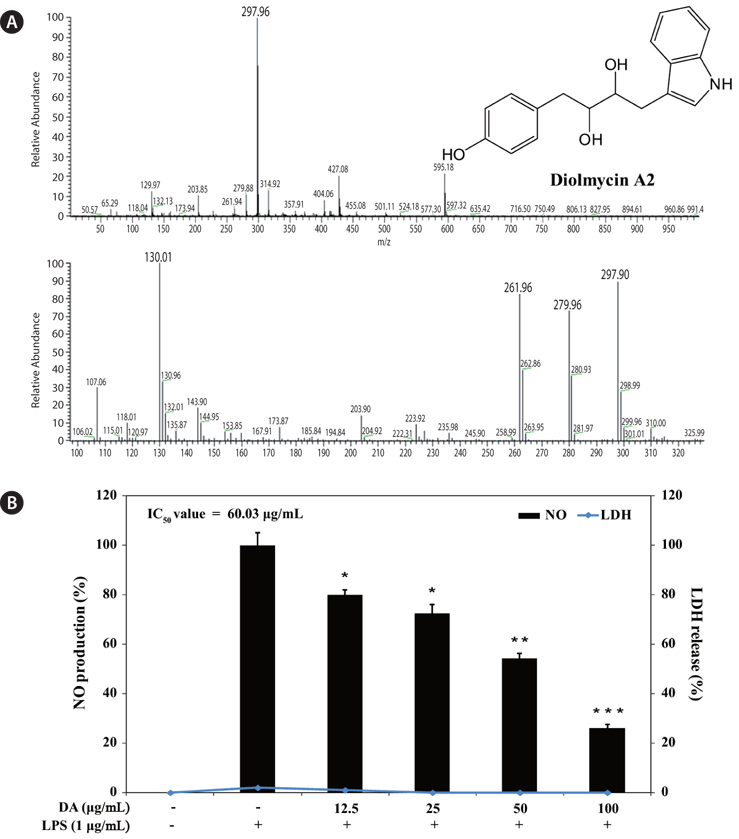
The
>
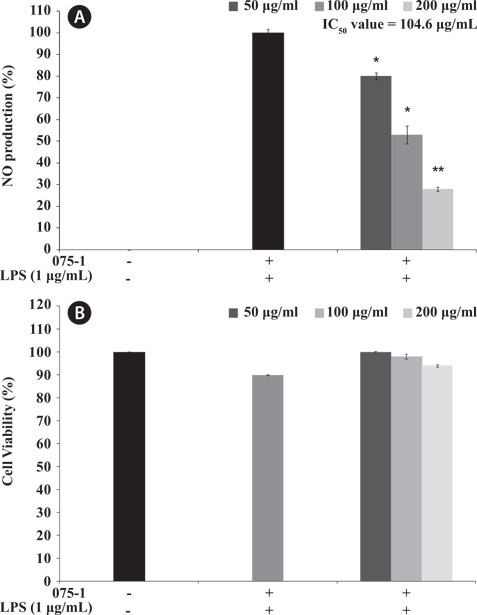
Inhibitory effect of diolmycin A2 on NO production in LPS-stimulated RAW 264.7 cells
To evaluate the inhibitory effect of diolmycin A2 on anti-inflammatory action, RAW 264.7 macrophages were stimulated with LPS (1 μg/mL) for 24 h to generate NO, and the accumulated NO in the culture medium was measured by the Griess reaction. The 50% inhibitory concentration (IC50) of diolmycin A2 was 60.03 μg/mL with no cytotoxicity on LPS-stimulated RAW 264.7 cells (Fig.5). Thus, diolmycin A2 strongly but dose-dependently inhibited NO production in LPS-induced RAW 264.7 cells.
Various secondary metabolites biosynthesized by bacteria inhabiting marine creatures have been shown to possess good bioactivity (Bringmann et al., 2003, 2005; Liu et al., 2005; Zheng et al., 2005; Pimentel-Elardo et al., 2010). Previous studies have shown that
Here, we investigated the anti-inflammatory activity of secondary metabolite extracts from various bacteria species based on NO produced in LPS-stimulated RAW 264.7 cells without cytotoxicity. In the combined results of both cell viability and NO inhibitory activity, the secondary metabolite extracts from
Overall, we confirmed the presence of biologically active compounds in