Autism Spectrum Disorder (ASD), which begins in infancy or before the first 3 years old, is neurodevelopmental disorders characterized by impaired social relatedness, and communication, repetitive behaviors, abnormal movement patterns, and sensory dysfunction.1-3 According to the most recent official estimates, the prevalence of ASD in the United States is a 0.91% of average.4 The incidence of ASD in Korea is also increasing, reported a prevalence of 2.64%.5 Therefore early detection and early treatment of autism education is important. Genetic factors causing ASD have been proposed,6 but genetic studies were carried out in a specific genetic background, to date, accurate early diagnosis of autism has not been able to develop the technology.
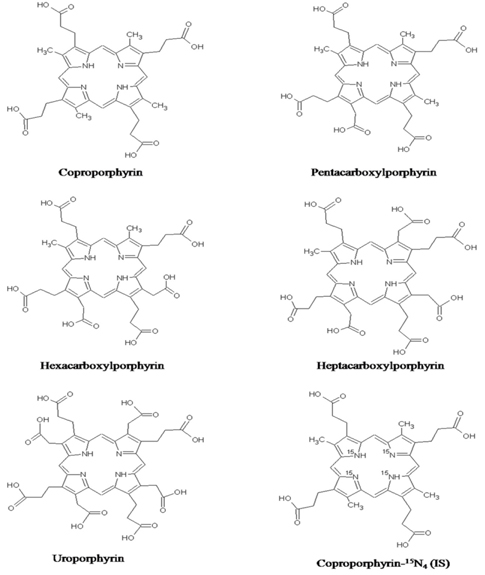
Porphyrins are intermediates of the heme biosynthetic pathway in mammalian tissues. All porphyrin molecules are consisted of cyclic tetrapyrrole (Figure 1). Quantitative analysis of porphyrins from plasma and urine is important because it might be a potential biomarker for ASD identification and classification.5
A number of techniques have been developed for determining urinary porphyrins by spectrofluorometry,6 high performance liquid chromatography,7,8 thin-layer chromatography,9 ion-pair high-performance liquid chromatography,10 capillary electrophoresis11 with fluorescence, matrix-assisted laser desorption ionization/time-of-flight mass spectrometry (MALDI-TOF),12 and fast atom bombardment mass spectrometry (FAB-MS).13 More recently, liquid chromatography-mass spectrometry (LC-MS), 14-16 and tandem mass spectrometry (LC-MS/MS)17-20 have been used to determine porphyrins in biological matrices (blood, urine and feces) because of their high sensitivity and specificity.
Although there have been many studies measuring urinary porphyrins by using LC-MS/MS, only a few studies have measured porphyrins levels in the plasma. And the validation of most of reported methods is still suffering from the interference of endogenous porphyrins. Endogenous porphyrins in the blank urine and plasma have been a problem for the validation of porphyrins analysis methods, and make it difficult to determine the limit of quantification. In this regard, using the porphyrin-free urine and plasma sample is appropriate to make the method reproducible.
In the present study, we have developed a rapid and sensitive liquid chromatography-electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) method for the simultaneous determination of porphyrins in human urine and plasma. Finally, the proposed method was successfully applied to clinical studies on the ASD diagnosis of 203 Korean children.
Coproporphyrin (CP, 97%), pentacarboxylporphyrin (PP, 90%), hexacarboxylporphyrin (HexaP, 90%), heptacarboxylporphyrin (HeptaP, 90%) and uroporphyrin (UP, 98.9%) were purchased from Frontier scientific Inc. (Logan, UT, USA). Coproporphyrin I-15N4 (IS, 98%), as an internal standard, was purchased from TRC Inc. (Toronto Research Chemicals, North York, Canada). HPLC grade acetonitrile and methanol were purchased from Burdick & Jackson (Muskegon, MI, USA). HPLC grade ammonium acetate, ethyl acetate, formic acid and active charcoal were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents were of analytical grade except those for HPLC.
>
LC-MS/MS operation conditions
A Shimadzu (Shimadzu corporation, Tokyo, Japan) LC-20 series LC system was equipped with degasser (DGU-20A3), binary pump (LC-20AD), column oven (CTO-20A), and autosampler (SIL-20AC) in which rack changer were used to inject 10 μL aliquots of the processed samples onto a Synergi Fusion RP column (150 mm × 2.0 mm, 4 μm, Phenomenex, Torrance, CA, USA). The column temperature was set at 30°C. The gradient mobile phase, with solvent A (0.1% formic acid in 2 mmol/L ammonium acetate, v/v) and solvent B (20% methanol in acetonitrile, v/v) was delivered to the column at a flow rate of 450 μL/ min. The gradient program started 10% mobile phase B, linear increase of B to 90% from 1.0 to 3.0 min. Mobile phase B was held at 90% for another 3.0 min (from 3.0 to 6.0 min) and then reduced linearly to the start condition (10% mobile phase B). The total run time was 15.0 min.
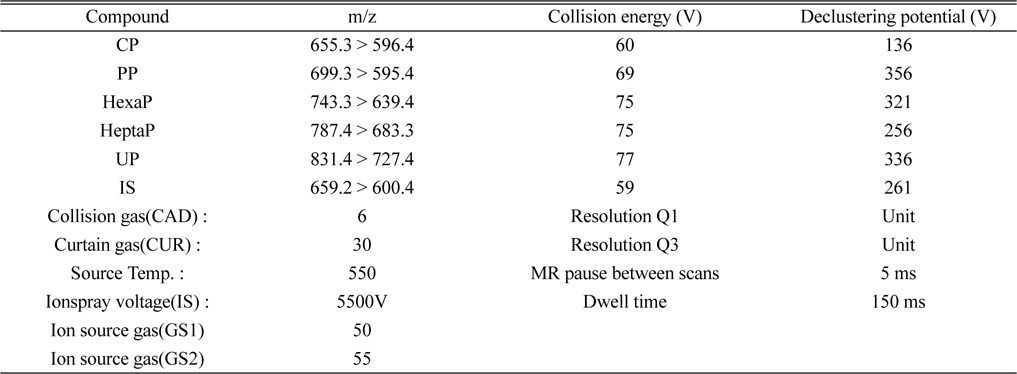
A MDS SCIEX (Foster City, CA, USA) API 5000 triple quadrupole tandem mass spectrometer was operated using an electrospray ionization source in the positive ion mode. The following parameters are listed in Table 1. Multiple reaction monitoring (MRM) of the precursor/product ion transitions were
[Table 1.] MRM conditions for the analysis of CP, PP, HexaP, HeptaP, UP and internal standard
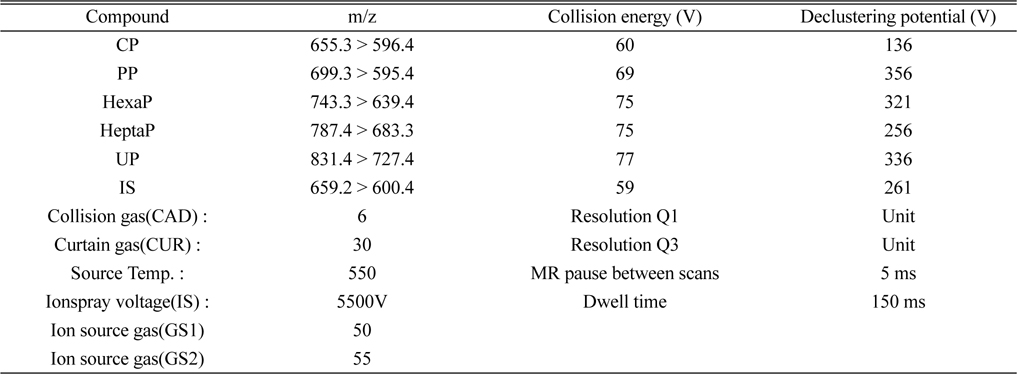
MRM conditions for the analysis of CP, PP, HexaP, HeptaP, UP and internal standard
>
Preparation of standard stock solutions and quality control (QC) samples
The stock solutions of five porphyrins (10 μmol/L) and the internal standard (10 μmol/L) were prepared in 6 mol/L formic acid and stored at 4°C. A series of working standard solutions of five porphyrins were prepared in a concentration range of 1-1000 nmol/L by diluting the stock solution with 1 mol/L formic acid. Internal standard stock solution was diluted with 50% methanol in 6 mol/L formic acid to a final concentration of 1000 nmol/L. Standard plasma samples were prepared by spiking 30 μL of working standard solutions into 270 μL of blank human plasma. Finally, standard plasma concentrations for the calibration curve were 0.1, 0.5, 1, 5, 10, 50 and 100 nmol/L. Quality control samples of 0.1, 0.3, 3, and 80 nmol/L were prepared for precision and accuracy of the studies.
>
Preparation of porphyrins-free urine and plasma
Pooled drug-free human urine and plasma were purified by using activated charcoal to remove endogenous porphyrins. Such porphyrins-free urine and plasma were used as blank biological matrix in this study. To be specific, 100 mL of the pooled blank matrix was mixed with 10 g of activated charcoal and the mixture was shaken moderately on an shaker overnight at room temperature. After centrifugation at 19,500 rpm for an hour, the supernatant of purified matrix was transferred to clean tubes and kept at ˗70°C until use.
A 300 μL urine sample was placed in a 2 mL eppendorf tube, and 10 μL internal standard working solution (1000 nmol/L) and 200 μL acetonitrile were added, and then the mixture was vortexed for 30 sec. After centrifugation at 13,000 rpm for 5 min, the supernatant was transferred into a 13 mL glass tube and evaporated to dryness under a stream of nitrogen at 40°C (MG2200, EYELA, Tokyo, Japan).
Prior to the extraction of plasma samples, 20 μL internal standard solution (1000 nmol/L) was added to 400 μL human plasma. Plasma samples were prepared by liquid-liquid extraction. For liquid-liquid extraction, 40 μL of 6 mol/L formic acid and 800 μL of ethyl acetate were added. After vortexing and subsequent centrifugation at 13,000 rpm for 5 min, the supernatant was transferred into a 13 mL glass tube. The organic layer was evaporated in a stream of nitrogen at 40°C.
All procedures were performed in dark places to prevent the photo-degradation of porphyrins. The dry residue of urine and plasma samples were reconstituted with 100 μL 1 mol/L formic acid and 10 μL aliquot was injected into the LC-MS/MS system.
Qualitative matrix effects were assessed by post-column infusion experiments, which identify whether the ion suppressions exist or not. The recovery was performed at 0.1 nmol/L concentration of analytes by comparing the area ratio with analyte and internal standard determined for the samples spiked with porphyrins before extraction, with the area of samples spiked post-extraction with porphyrins at the same concentration. The selectivity of the method was investigated by analyzing at least six different lots of blank human urine and plasma. Double blank samples (processed without internal standard) and blank samples (processed with internal standard only) were prepared.
Calibration curves were constructed by the peak area ratio of each porphyrin to internal standard against the analyte concentration. The acceptance criterion for each back-calculated standard concentration was ±15% standard deviation (SD) from the nominal value except at the limit of quantification (LOQ), where it should not exceed ±20%. The LOQ were defined as the minimum concentration with a signal-to-noise ratio of 10.
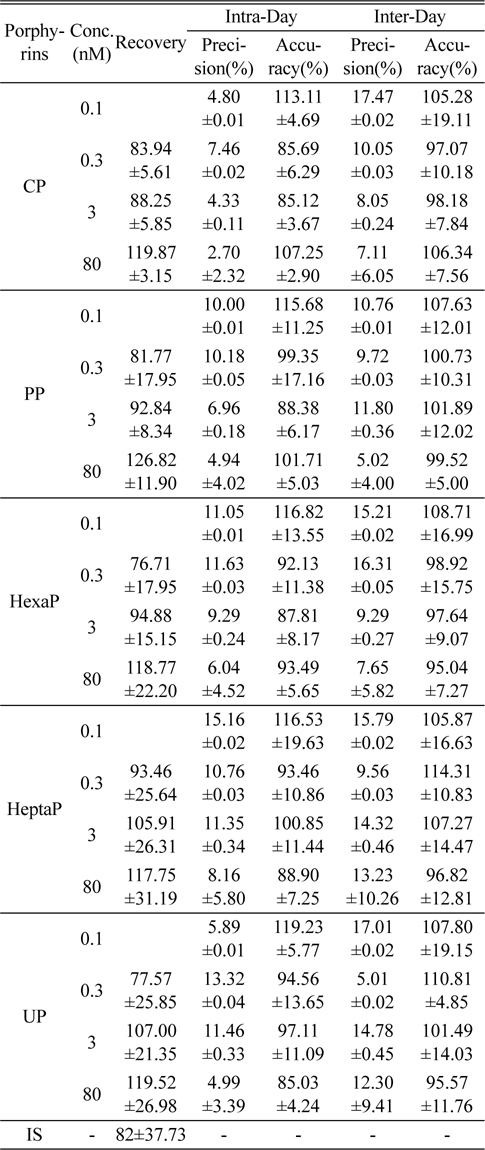
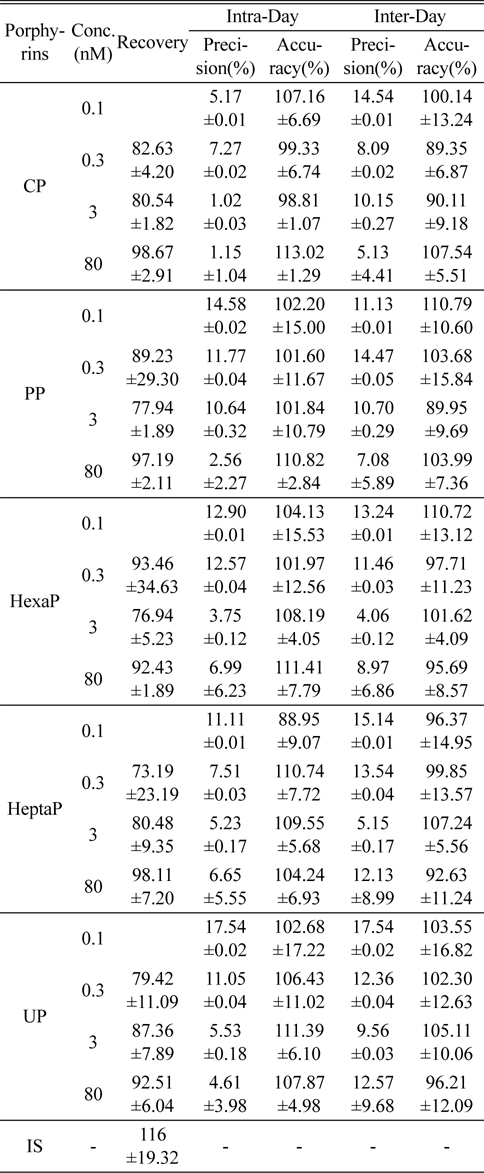
The intra-day precision and accuracy were estimated by analyzing five replicates (n=5) at four different concentration levels, 0.1, 0.3, 3, and 80 nmol/L. The inter-day precision and accuracy were determined by analyzing the four different concentration samples (n=5). Accuracy was expressed as the percentage of observed value to true value, and precision was expressed as the relative standard deviation (RSD). The criteria for acceptability of the data included accuracy below ±15% deviation from the nominal values and precision below ±15% RSD, except for the LOQ.
Short-term stability of the analytes in human urine and plasma was performed over 12 h at room temperature using two different QC samples (0.3 and 80 nmol/L). Freeze-thaw stability through three freeze-thaw cycles was assessed using two different QC samples. The aliquots of samples, which were stored at ˗70 ± 5°C, were thawed completely at room temperature, and then the samples were refrozen in the freezer (three freeze-thaw cycles). The autosampler stability was determined by injecting replicate preparations of processed QC samples into the autosampler at 4°C for up to 24 h after the initial injection. All stability tests were estimated by analyzing three replicates. Samples were considered to be stable if assay values were within the acceptable limits of accuracy (±15% deviation) and precision (±15% RSD).
The study protocol was approved by Institutional Review Board (IRB) from International Scientific Standards, Inc. (Chuncheon, Korea). Clinical studies were carried out in Rudolph (the Korea institute for children's social development), and participants were divided into autistic group (patient) and the control group (healthy human). Medical laboratory test was conducted in Institute for EONE Life Science Metabolomics Research Center (Incheon, Korea). Along with study samples, QC samples at low, medium, and high concentration were assayed in duplicate and were distributed among unknown samples in the analytical run.
>
Preparation of porphyrins-free blank plasma and urine
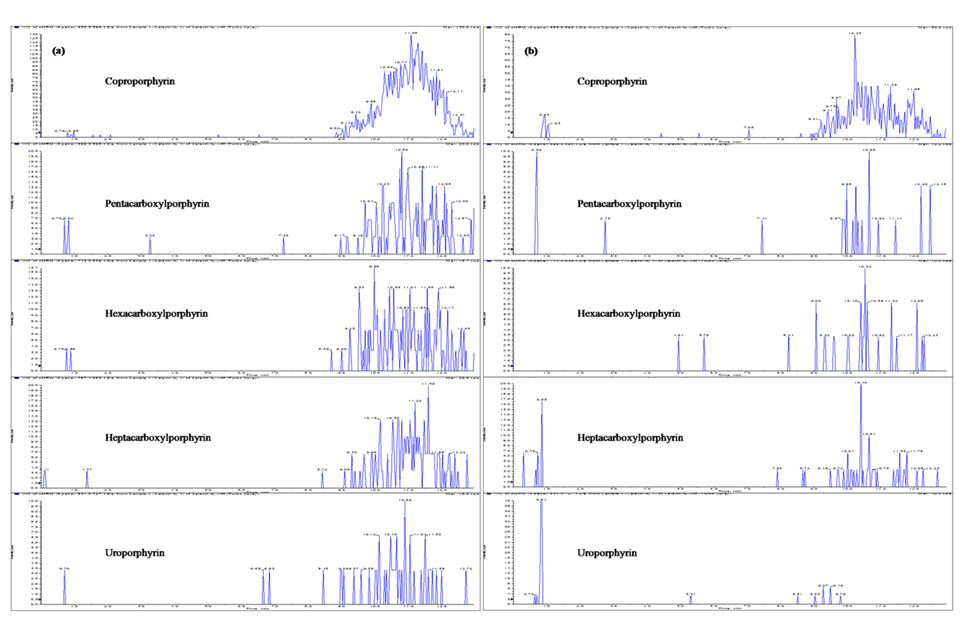
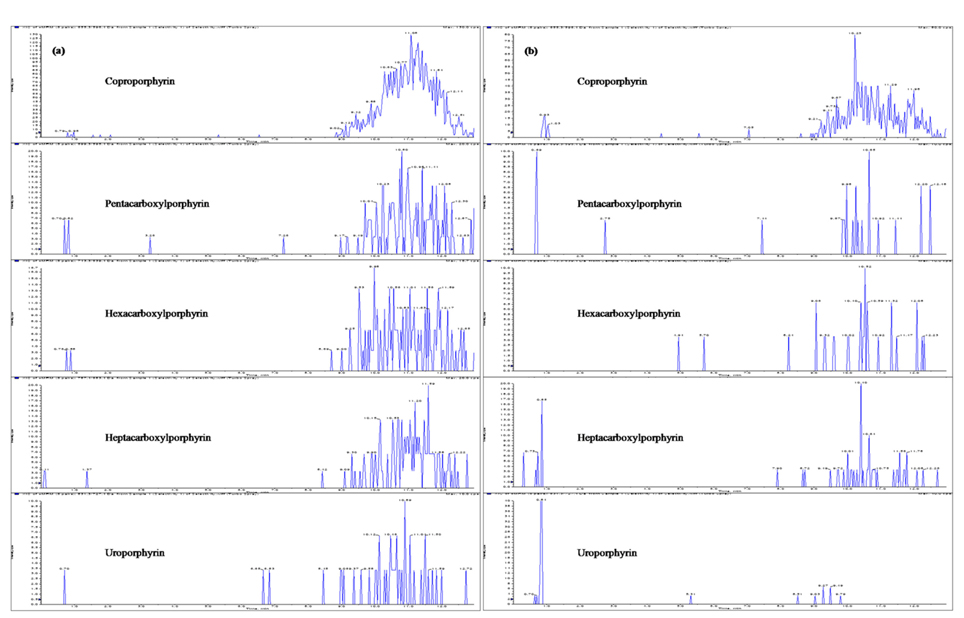
Until now, most reported analytical methods for porphyrins are not free from the interference of endogenous porphyrins in human urine or plasma. Endogenous porphyrins have been a problem for porphyrins analysis, and make it difficult to determine the limit of quantif-ication. Generally, control plasma has been processed with activated charcoal to eliminate free, and low molecular weight hormones.21 In this study, activated charcoal was used to remove the endogenous porphyrins from the pooled urine and plasma. Our results demonstrated that endogenous porphyrins were efficiently removed by adsor-ption to activated charcoal from human plasma and urine, as shown in Figure 2. This kind of porphyrins-free plasma and urine were very suitable to the blank matrices for preparation of calibration standard and quality control samples.
>
Optimization of HPLC and MS/MS spectrometry
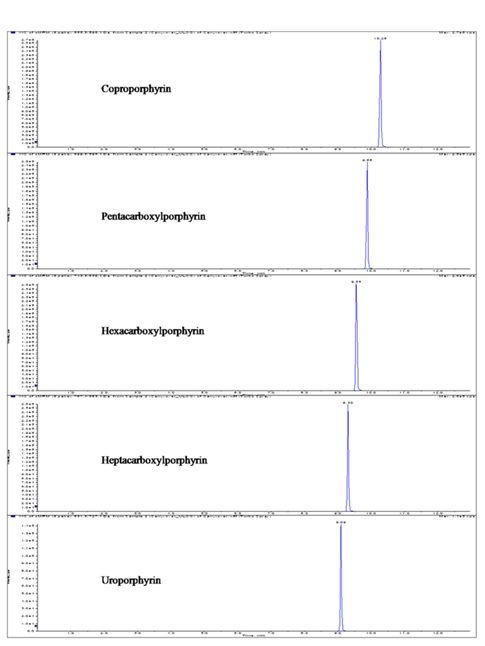
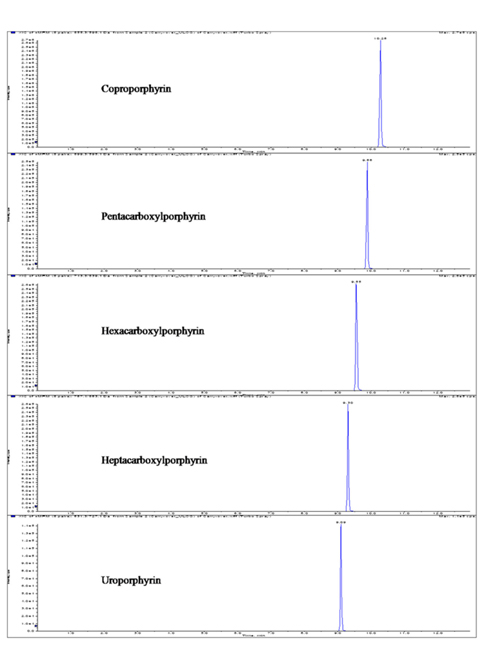
We have focused on manipulating chromatographic parameters to increase the intensity and achieve fast separation of porphyrins with high chromatographic resolution. Figure 3 shows the reconstructed LC-ESI-MS/MS MRM chromatograms of standard mixture of the five porphyrins. The five porphyrins were further characterized by their MS/MS product ion spectra. Previous studies have shown the major fragmentation pathway of porphyrins.22
In the present study, several columns, including Synergi Fusion RP (150 mm × 2.0 mm, 4 μm), Hypersil Gold (150 mm × 2.1 mm, 5 um, Thermo Fisher Scientific, Pittsburgh, PA, USA), and Halo C18 (100 mm × 4.6 mm, 2.7 um, Advanced Material Technology, Wilmington, DE, USA), were compared on the basis of peak separation and sharpness in order to evaluate column performance (data not shown). Our results showed that the Synergi Fusion RP column exhibited the most satisfactory performance in chromatographic separation and sensitivity. Thus, this column was selected as the stationary phase for the determination of five porphyrins in human urine and plasma.
To obtain complete separation and rapid analysis of five porphyrins, several types of buffer solution with an organic solvent in different proportions have been tried as a mobile phase. Initially, we used the isocratic mode with flow rate of 0.45 mL/min, but there was a broad and some peak tailing with poor resolution. Ultimately, the optimal mobile phase, which improved peak shape and separation, was consisted of solvent A (0.1% formic acid in 2 mmol/L ammonium acetate, v/v) and solvent B (20% methanol in acetonitrile, v/v) with gradient elution. The total run time was 15 min.
The HPLC system was coupled with triple quadrupole mass spectrometer (API 5000TM; Applied Biosystems). Much higher ion intensities of porphyrins and internal standard were observed in positive of ESI ion mode. The mass spectrometric parameters, including desolvation temperature, curtain gas, collision energy (CE), nebulizing gas (GS1), declustering potential (DP), collision cell exit potential (CXP), and entrance potential (EP) were optimized to obtain the highest signal responses for the precursor and product ions of porphyrins and internal standard.
Figure 2 shows the LC-MS/MS chromatograms of (a) blank human urine and (b) blank human plasma. No interfering peaks from endogenous components were detected at the retention times of porphyrins and internal standard. The retention times of CP, PP, HexaP, HeptaP, UP and internal standard were 10.3, 9.9, 9.6, 9.3, 9.1 and 10.1 min, respectively. The total chromatographic run time was 15 min (Figure 3). The post-column infusion method was used in this validation. There was no ion suppression at the region for the retention times of porphyrins and IS. The high recoveries proved that the method was valid for the determination of five porphyrins in urine and plasma samples (Table 2 and 3). The standard calibration curves for five porphyrins were constructed using 7 points calibration standard solutions (0.1-100 nmol/L). The coefficients of determination (r2) were greater than 0.99. Table 2 and 3 show the results for intra- and inter-day precision and accuracy. The limit of quantification (LOQ) was determined as the concentration at which the signal-to-noise (S/N) ratio was greater than 10, precision of < 20% and accuracy between 80-120% for both intra- and interday assays. In this study, the LOQs for five porphyrins were all 0.1 nmol/L.
[Table 2.] Recovery, precision and accuracy for the determination of porphyrins in human urine (n=5)
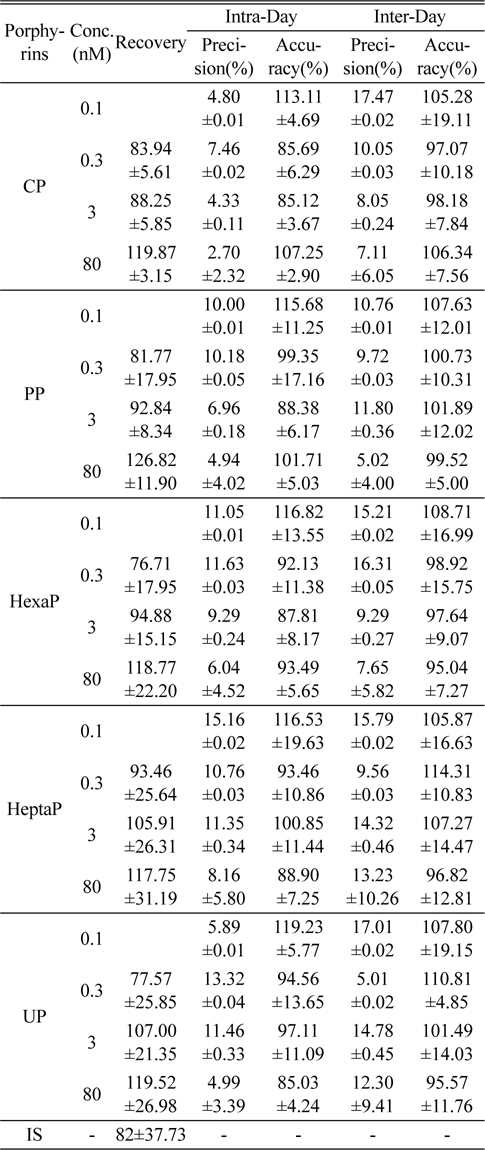
Recovery, precision and accuracy for the determination of porphyrins in human urine (n=5)
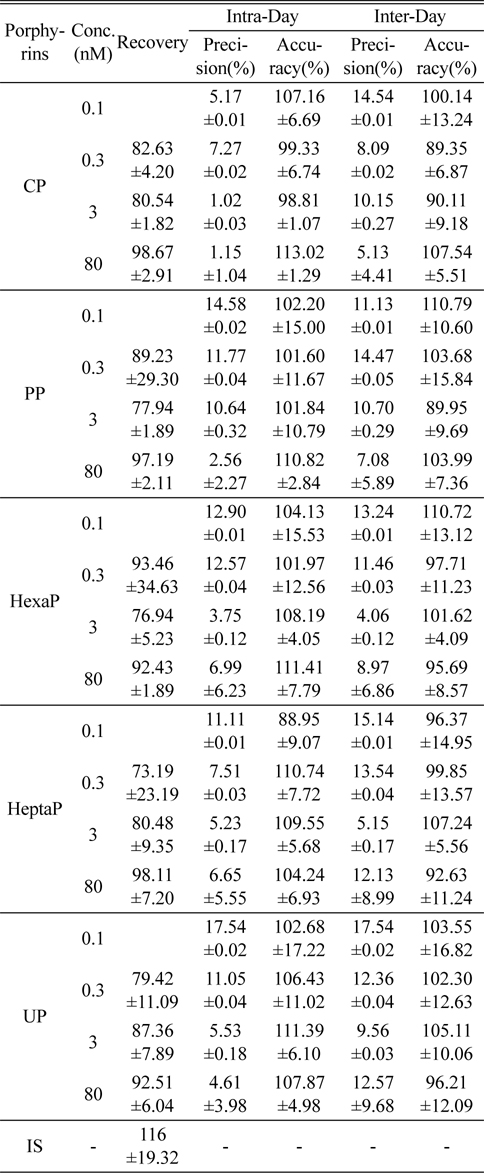
Recovery, precision and accuracy for the determination of porphyrins in human plasma (n=5)
The results of freeze-thaw, short-term, and autosampler stability test for porphyrins are shown in supplementary Table S1. These results indicate that porphyrins and internal standard are stable after three freeze-thaw cycles (˗80°C to room temperature), over 12 h of storage at 25°C, and over 23 h at 4°C in the autosampler chamber as a processed sample solution. The long-term stability of porphyrins in human urine was 4 weeks storage at ˗20°C and 1 week storage at ˗20°C reported previously.23,24
>
Application of the method to a clinical study
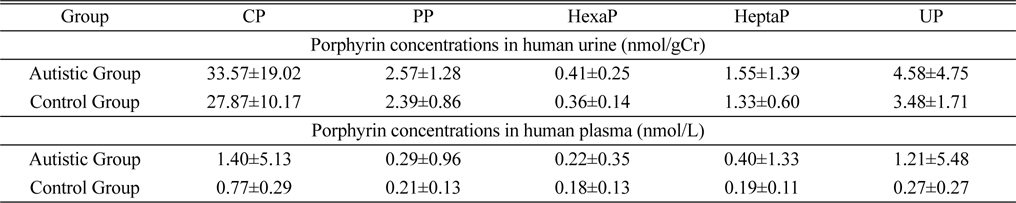
The study subjects included 102 healthy persons and 101 ASD patients. The method was successfully applied to determine the endogenous porphyrins in human urine and plasma. The measured concentrations of porphyrins from 203 Korean children are shown in Table 4. A similar pattern was observed in both urine and plasma samples. The mean concentrations of porphyrins in autistic group were higher than those of healthy group.
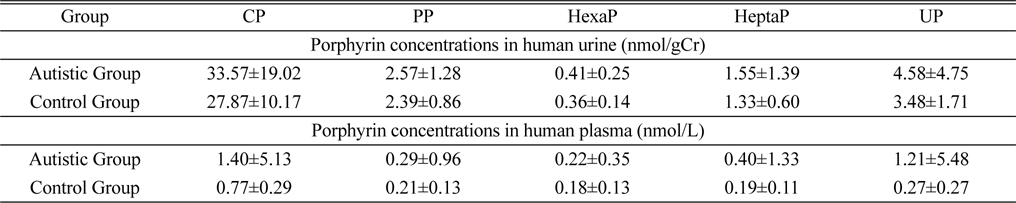
Porphyrins concentrations in human urine and plasma samples from 203 Korean children (mean±SD)
A selective and sensitive LC-MS/MS method for analysis of five porphyrins in human urine and plasma samples was developed and validated. The activated charcoal was used to remove the endogenous porphyrins from the pooled human urine and plasma for reducing interference. The method was successfully applied to determine plasma and urinary porphyrins from clinical samples as well.