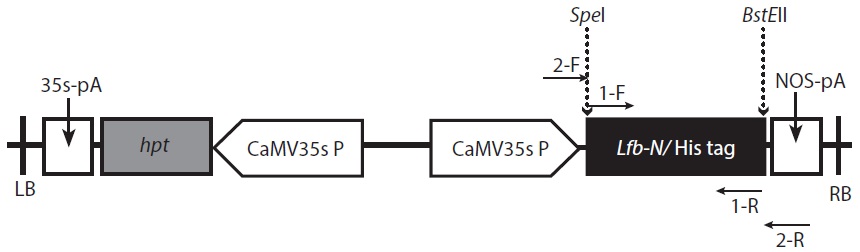
The purpose of this study was to express bovine lactoferrin N-lobe in Chlorella vulgaris, a green microalga, using the pCAMBIA1304 vector. Chlorella-codon-optimized bovine lactoferrin N-lobe (Lfb-N gene) was cloned in the expression vector pCAMBIA1304, creating the plasmid pCAMLfb-N. pCAMLfb-N was then introduced into C. vulgaris by electrotransformation. Transformants were separated from BG-11 plates containing 20 μg mL-1 hygromycin. Polymerase chain reaction was used to screen transformants harboring Lfb-N gene. Finally, total soluble protein was extracted from the transformants, and the expression of Lfb-N protein was detected using western blotting. Using this method, we successfully expressed bovine lactoferrin in C. vulgaris. Therefore, our results suggested that recombinant lactoferrin N-lobe, which has many uses in the biomedical and pharmaceutical industries, can be produced economically.
Lactoferrin (also called lactotransferrin) is a member of the transferrin family of proteins (Aisen and Listowski 1980, Brock 1985, Rose et al. 1986) and is found in milk, saliva, and tears. Lactoferrin is secreted from mammary glands, mucous, and secondary granules of neutrophils, which function in the nonimmunological defense system. Moreover, bovine colostrum contains 5 mg mL-1 lactoferrin, while normal milk contains only about 0.02 mg mL-1 lactoferrin. Human lactoferrin consists of 691 amino acids, which associate with 2 glycan chains of glycoproteins (Metz-Boutigue et al. 1984), and bovine lactoferrin contains 689 amino acids (Picerce et al. 1991), with a predicted molecular weight of about 76 kDa, or 83 kDa when accounting for its glycoconjugates. The lactoferrin glycoprotein consists of 2 lobes, with each lobe having an Fe3+ binding site. The antibacterial core of lactoferrin, called lactoferricin, can be produced through pepsin-mediated cleavage. This peptide has antimicrobial properties and is therefore a potential therapeutic agent in various clinical settings.
Currently, most commercially obtainable recombinant protein is produced using bacteria, yeast fungi, or bioreactors designed for animal cell culture. Bacterial protein expression systems lack critical post-transcriptional and post-translational regulatory mechanisms, such as splicing, glycosylation, and protein assembly. Moreover, there is a high possibility that purified proteins may contain bacterial endotoxin and protease contaminants. Similarly, yeast expression systems, while eukaryotic in nature, do not have machinery for the glycosylation. Currently, most proteins generated for medical use are cultured and produced in animal cells. However, it is difficult to mass produce such proteins, the cost of culture medium is high, and cultures are easily contaminated. Moreover, low yields have high costs, making this type of expression system unsustainable for clinical applications. Additionally, while plant-based expression systems have many advantages for mass production of stable, functional proteins, challenges to these methods include environmental pollution by genetically modified plants, allergic reactions to plant components, contamination of protein, and regulation of medical protein permission (Choi and Sim 2009).
Genetic engineering technology has been under development for more than ten years now, and eukaryotic microalga represent an alternative expression system to overcome issues of other expression systems based in animals, fungi, bacteria, and plants. Recently, many studies have investigated the production of antibodies, medical proteins, and vaccines in microalgae (Yang et al. 2006, Tran et al. 2009, Dreesen et al. 2010). Moreover, microalgae have the potential for mass culture in growth conditions similar to that required for general crops. For these reasons, microalgae have become the focus of novel gene manipulation methods to increase the productivity of natural components and novel compound. However, while studies are currently being conducting to investigate the potential for genetic transformation in microalga models, such as
In the present study, to the best of our knowledge, we report the first successful expression of bovine lactoferrin N-lobe gene in
>
Codon optimization and the expression vector
In this study, bovine lactoferrin N-lobe (including a lactoferricin) was used for codon optimization and determined to be
>
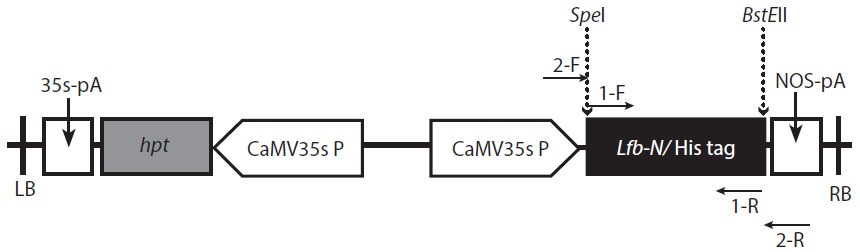
Construction of the expression vector
The vector construction was carried out as described by Ebrahimi et al. (2012), with some modifications. For construction of the expression vector, the purified pLfb-N vector and the pCAMBIA1304 vector were digested with
Electrotransformation was carried out as described by Wang et al. (2007
electrotransformation was performed by electroporator ECM 2001 (BTX, Holliston, MA, USA) under voltage 1,000 V at 25 μF and 200 ohm to generate the transgenic
>
Genomic DNA extraction and screening of putative transgenic Chlorella vulgaris DNA by PCR
Extraction of
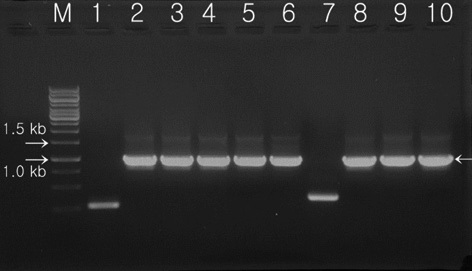
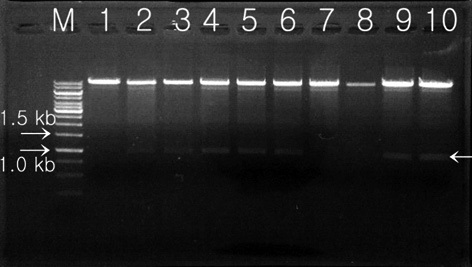
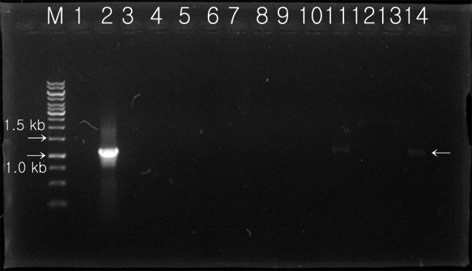
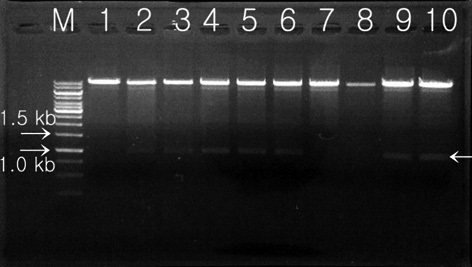
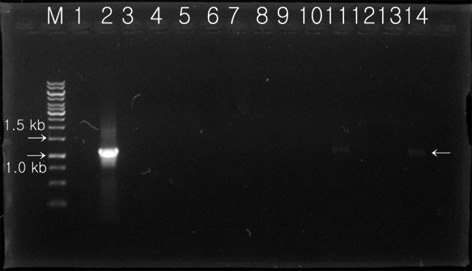
Transformed colonies was randomly selected and cultured for 7 days in 5 mL BG-11 broth (containing 20 mg L-1 hygromycin) at 25℃, with shaking at 180 rpm. Light conditions were set at 50 μmol m-2 s-1 using a fluorescent light for 14 h, with a 10 h night cycles. Cultured transformants were recovered after processing with a centrifugal separator, and extracted genomic DNA was used as a temple for PCR. Two oligonucleotide primers were synthesized for detection of the transferred Lfb-N DNA fragment by PCR analysis. PCR analysis was performed with the primer pair 2-F (5′-GAGAACACGGGGGACTCTTG-3′) and 2-R (5′-GGGGAAATTCGAGCTGGTCA-3′) specific to the
>
Protein extraction and western blot analysis
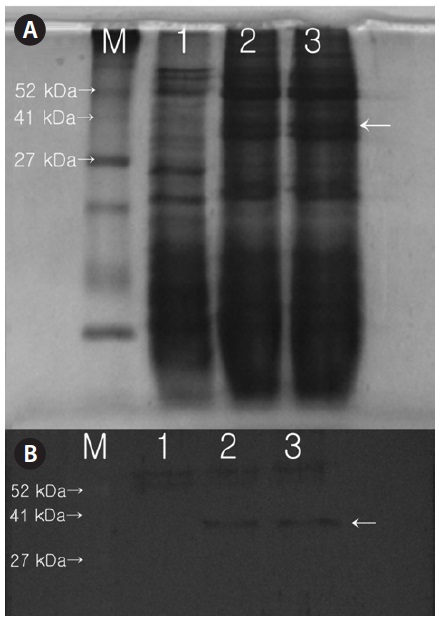
A 50-mL sample of cell suspension (108 cells mL-1) was centrifuged for 5 min at 2,000 ×
>
Codon optimization and construction of the expression vector pCAMLfb-N
In this study, we optimized the
The digested Lfb-N was cloned between 3′ end of CaMV35s and 5′ end of the
>
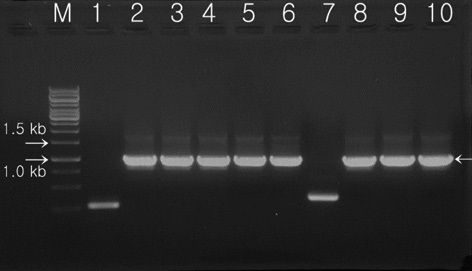
Electrotransformation and screening of putative Chlorella vulgaris by PCR analysis
The pCAMLfb-N was used to transform
hygromycin-resistant single colonies, confirmed within 7 days on selection media, were randomly selected, grown in liquid media before the DNA was extracted, and used in PCR analysis. The primers 2-F and 2-R were used to carry out the PCR amplification for screening of
>
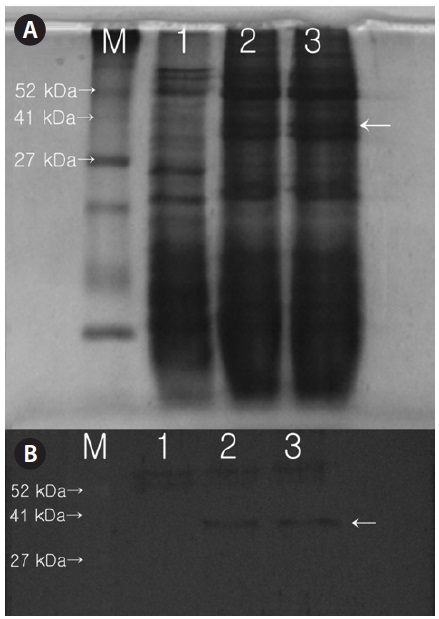
Protein expression and western blot analysis
Expression of the protein product from the introduced
In microalgae, the expression of heterologous proteins presents several difficulties. First, microalgae have an unusual GC-rich codon bias in their nuclear genes; therefore, codon optimization must be performed on any gene for which high levels of protein expression are desired (Heitzer et al. 2007). Additionally, expression levels of optimized foreign genes may vary considerably due to position effect that is driven by random integration of the gene of interest and strong silencing mechanism that drives by epigenetic phenomena similar to those in land plants (Schroda 2006). In
as in land plants, silenced multiple-copy transgenes exhibit high levels of DNA methylation (Cerutti et al. 1997, Babinger et al. 2001). In contrast, single-copy transgenes are subject to transgene silencing without detectable cytosine methylation (Cerutti et al. 1997).
The nuclear genome and the plastid genome have highly divergent codon usage, with the chloroplast preferring an A or T in the wobble position, while the nuclear genome prefers a G or C (Nakamura et al. 2000). Using GFP, early work showed that codon optimization to reflect the genome bias could increase transgene protein accumulation 5-fold in the nucleus (Fuhrmann et al. 1999) and up to 80-fold in the chloroplast (Franklin et al. 2002). Today, recombinant genes are universally codon optimized for improved protein expression in almost every system (Puigbo et al. 2007, Xia 2007, Puigbo et al. 2008
Various genetic transformation methods have been used to express recombinant proteins through introduction of genes into microalgae like
In this study, we used electrotransformation to move recombinant DNA into microalgae, allowing the growth of hygromycin-resistant colonies. The effectiveness of microalgal electroporation, or the induction of macromolecular uptake by exposing cell walls to high intensity electrical field pulses, was first reported by Brown et al. (1991). Electroporation specifically disrupts lipid bilayers, leading to efficient molecular transport across the plasma membrane (Azencott et al. 2007). Efficient electrotransformation was achieved in both wild-type and cell wall-deficient strains (Brown et al. 1991). The transformation efficiency of electroporation is two orders of magnitude higher than the glass beads method, and only requires relatively simple equipment (Shimogawara et al. 1998). Important parameters affecting the effectiveness of electroporation include field strength, pulse length, medium composition, temperature and membrane characteristics (Brown et al. 1991) as well as the concentration of DNA (Wang et al. 2007
>
Expression of recombinant lactoferrin
Studies of recombinant lactoferrin expression have been conducted in several hosts, including
Thus, genetic transformation in a plant-based expression system has some benefits, enabling the production of stable, functional protein. However, mass culture via such plant-based systems is limited due to spatial restrictions and risks of exposure of genetically transformed organisms into the ecosystem. However, as we have shown, recombinant protein production using microalgae can be performed using micro-organism culture within a bioreactor, the volume of the culture is adjustable, and the risk of exposure to the ecosystem is relatively low.
In conclusion, we confirmed that recombinant bovine lactoferrin N-lobe could be expressed from the microalga