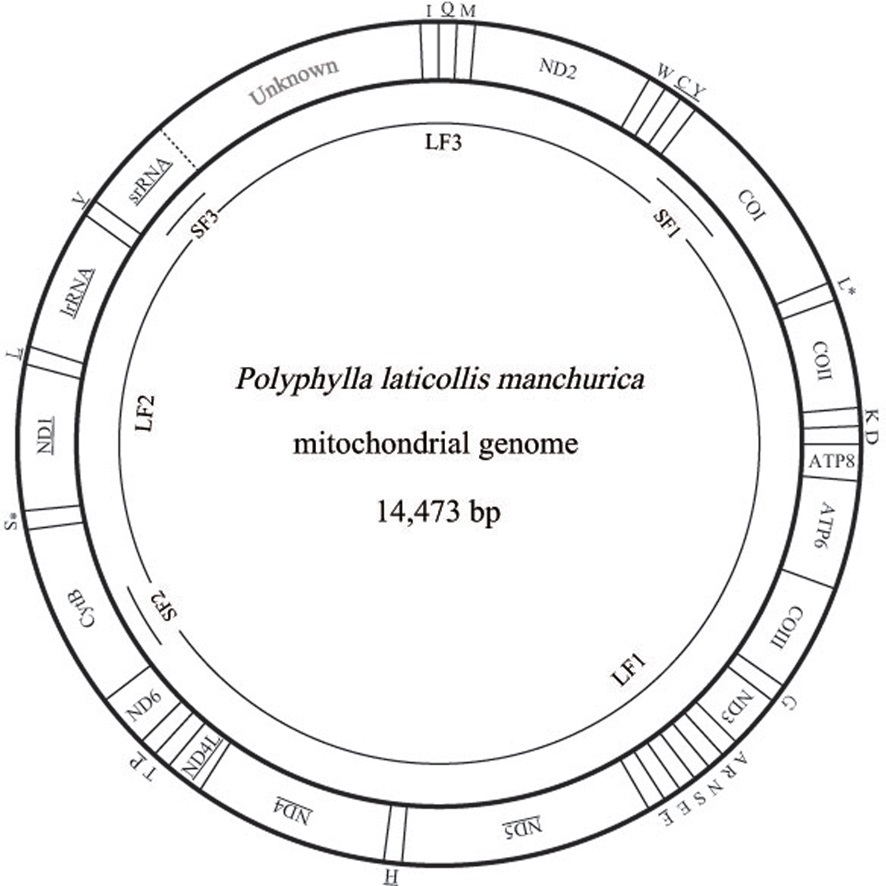
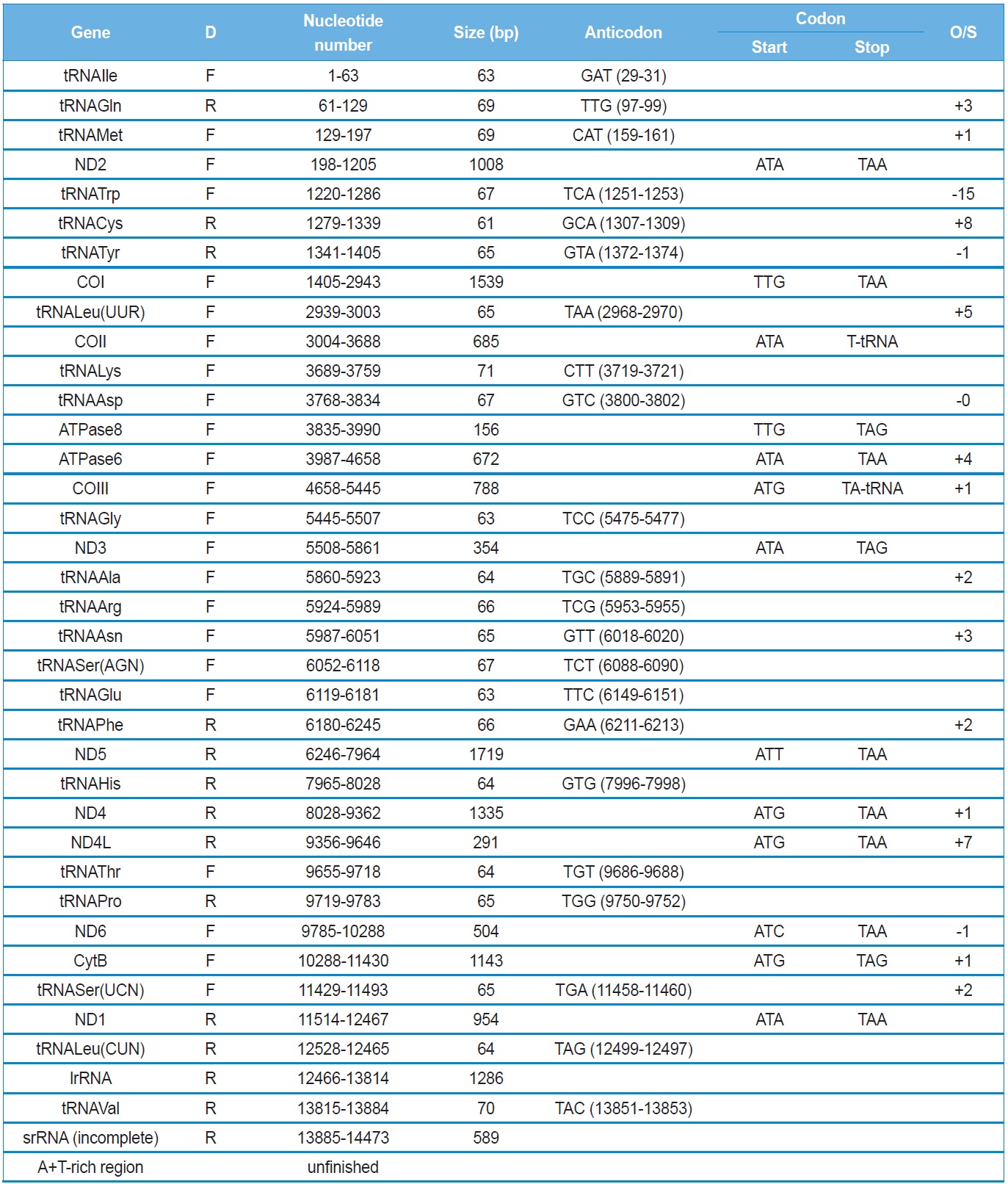
In this study, we present the nearly complete mitogenome sequences of the garden chafer, Polyphylla laticollis manchurica, which is listed as an endangered species in Korea. The P. l. manchurica mitogenome, which includes unfinished whole A+T-rich region and a partial srRNA was 14,473-bp long, possessing typical sets of genes (13 PCGs, 22 tRNA genes, and 2 rRNA genes). Gene arrangement of the P. l. manchurica mitogenome was identical to the common one found in the majority of insects. The 5 bp-long motif sequence (TAGTA) that has been suggested to be the possible binding site for the transcription termination peptide for the major-strand was also found in the P. l. manchurica mitogenome between tRNASer(UCN) and ND1. The start codon for COI gene and ATPase8 was designated as a typical TTG. All tRNAs of the P. l. manchurica showed a stable canonical clover-leaf structure of other mt tRNAs, except for tRNASer(AGN), DHU arm of which could not form stable stemloop structure. As has been previously determined, the high A/T content was unanimously observed in P. l. manchurica in terms of A/T bias in the third codon position (73.5%) compared with the first (66.4%) and second codon position (66.2%). The PCGs encoded in major-strands are slightly T-skewed, whereas those of the minor-strand are A-skewed, indicating strand asymmetry in nucleotide composition in the Coleoptera including P. l. manchurica.
Animal mitochondrial genomes (mitogenomes) are approximately 16~20 kbp, and are encoded with a remarkably conserved set of 37 genes: 13 protein-coding genes (PCGs), two ribosomal RNA (rRNA) genes, and 22 transfer RNA (tRNA) genes, and one major non-coding sequence, which is termed the control region (Boore, 1999). This control region in insect instead is called as the A+T-rich region due to the high adenine and thymine (A/T) content, and in fact, this region contains the highest A/T content of any region of the mitogenomes in insects (Kim
The mitogenome information has greatly been devoted to our understanding of several fields of biology (
Up to now, more than 250 mitogenome sequences have been determined from a variety of insects, but this list includes only ~34 coleopteran species (http://www.ncbi.nlm.nih.gov/genomes/ORGANELLES). Considering that the suborder Polyphaga contains the vast majority of beetle diversity, with at least 300,000 described species (90% of the beetles) belonging to more than 100 families in four infraorders (Hammond, 1992), genomic information is extremely limited. In particular, the complete mitogenome sequence of the infraorder Scarabaeiformia in Polyphaga is available only for two species (Cameron
In this paper, we report the mitogenome sequence of
>
Specimen collection and genomic DNA extraction
The garden chafer,
>
PCR amplification, cloning, and sequencing
Three short fragments, corresponding each about 500~700 bp were amplified from three genes, such as COI (SF1), CytB (SF2), and srRNA (SF3) (Fig. 1). Primers for these
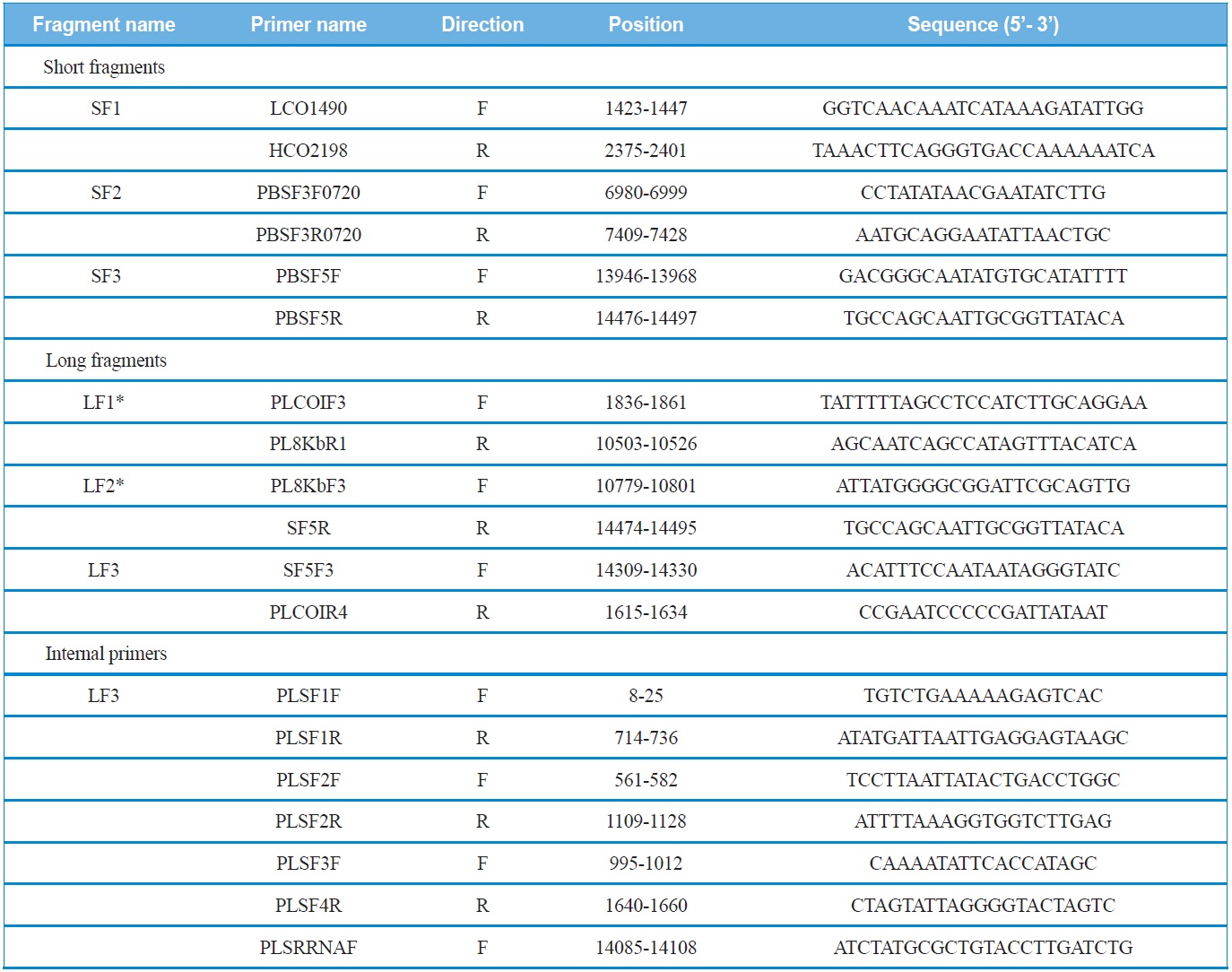
List of primers used to amplify and sequence the mitochondrial genomes of Polyphylla laticollis manchurica
short fragments were designed via the alignment of several coleopteran mitogenomes sequenced in their entirety. These short fragments were amplified with AccuPower® PCR PreMix (Bioneer, Korea) using an initial denaturation at 94℃ for 4 min, followed by 35 cycles of 30 s denaturation at 94℃, 40 sec annealing at 50-55℃, and a 60 sec extension at 72℃. The final extension step was continued for 8 min.
Using the sequence information obtained from the short fragments, three primer sets specific to each species were designed to amplify three long fragments (LF1 ~ LF3) which overlapped with the short fragments (SF1 ~ SF3). PCR cycles were as follows: denaturation for 2 min at 96℃, followed by 30 cycles of 10 sec at 98℃ and 15 min at 58-65℃, and a final 10 min extension at 72℃. In order to sequence the long fragments, both primer walking and shotgun approaches were used, because the success of sequencing varied depending on fragments. Nevertheless, both approaches were unsuccessful for LF3, and, thus, the partial LF3 that encompasses the 5’-end of the srRNA and the whole A+T-rich region was unfinished. We believe this may have happened because this region is exceptionally long, containing unexpectedly long repeat regions and a high A/T content, considering previous sequence results of other coleopteran species, such as
For the primer walking method, internal primers were directly used to complete the sequences of the long fragment subsequent to purification with OIAquick PCR Purification Kit (Qiagen, USA). For the shotgun approach the long PCR fragments were subjected to shearing into 1~5 kb fragments (Gene Machine, USA) and were cloned into the pUC118 vector (Takara Biomedical, Japan). Each resultant plasmid DNA was isolated using a Wizard Plus SV Minipreps DNA Purification System (Promega, USA). DNA sequencing was conducted using the ABI PRISM® BigDye® Terminator v3.1 Cycle Sequencing Kit and the ABI PRISM® 3100 Genetic Analyzer (PE Applied Biosystems, USA).
>
Gene identification and structure
The boundary of individual thirteen mitochondrial protein-coding genes (PCGs) and individual two rRNAs were determined through the alignment of the homologous sequences of known full-length coleopteran mitochondrial genome sequences using the CLUSTAL X program (Thompson
>
Comparative mitochondrial gene analyses
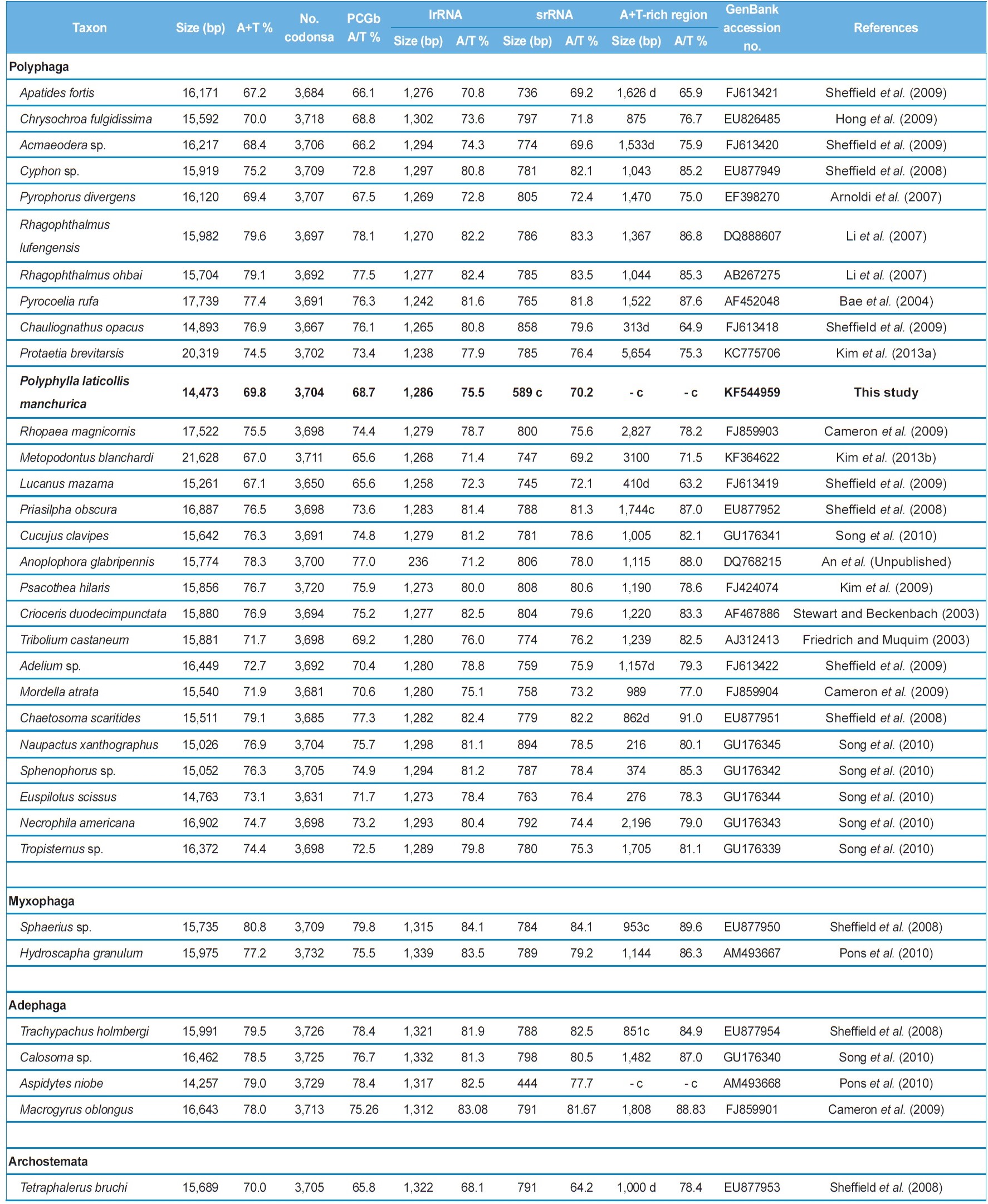
We compared 35 species of coleopteran insects including the currently sequenced
The
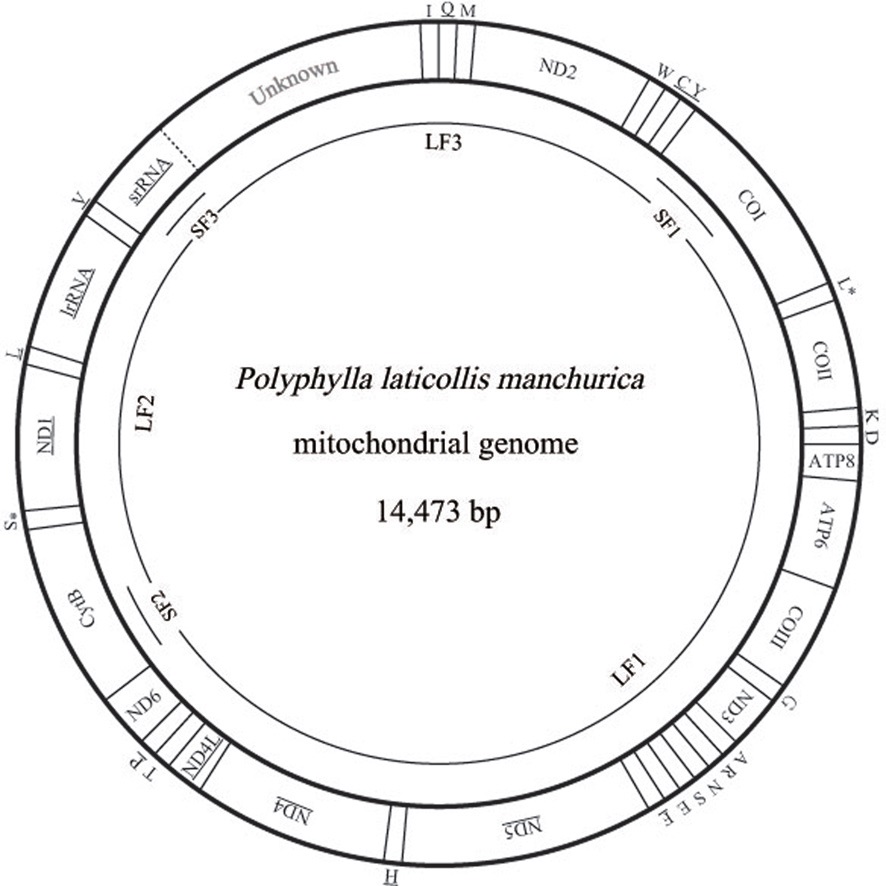
[Table 2.] Summary of the mitochondrial genome of Polyphylla laticollis manchurica
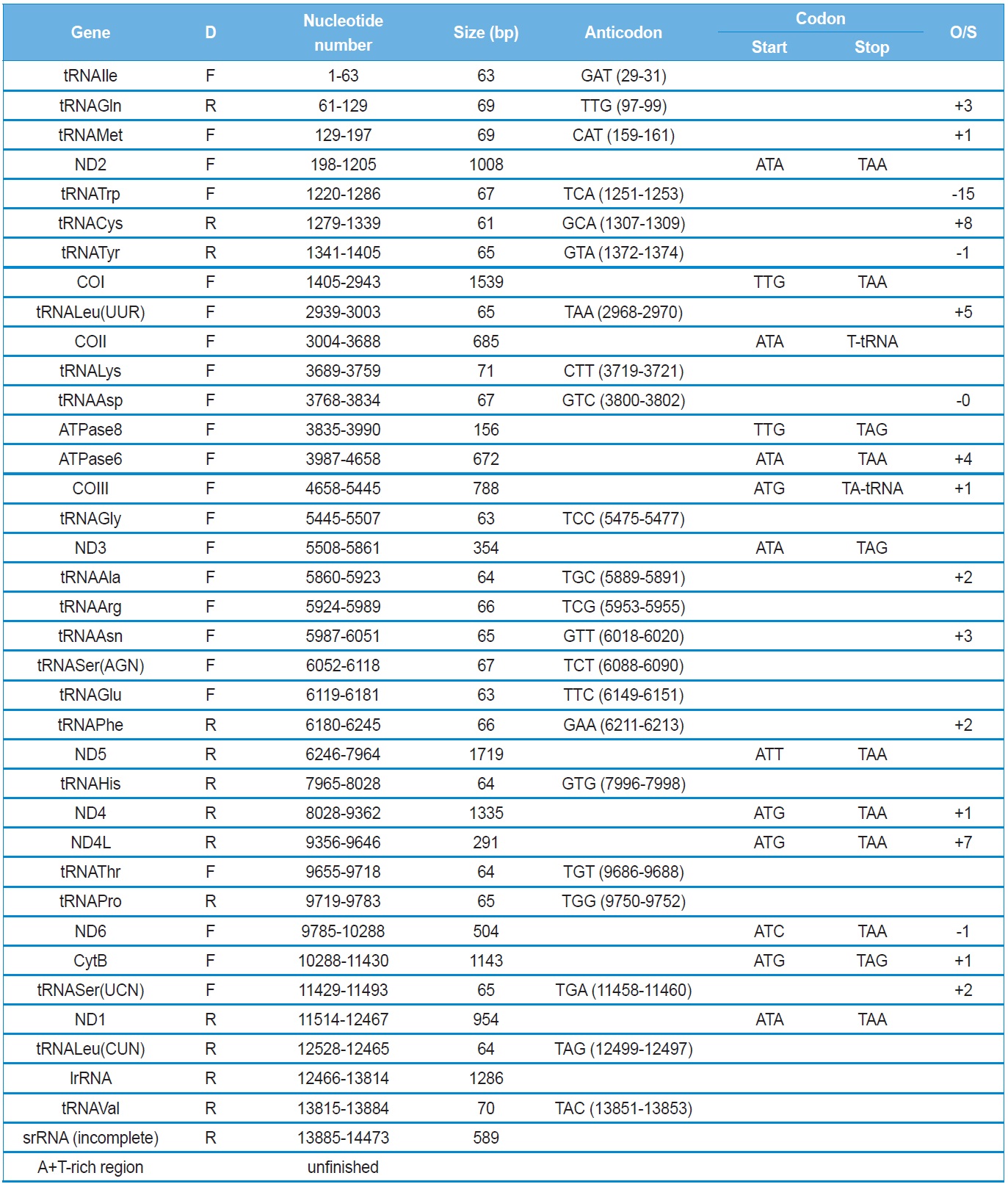
Summary of the mitochondrial genome of Polyphylla laticollis manchurica
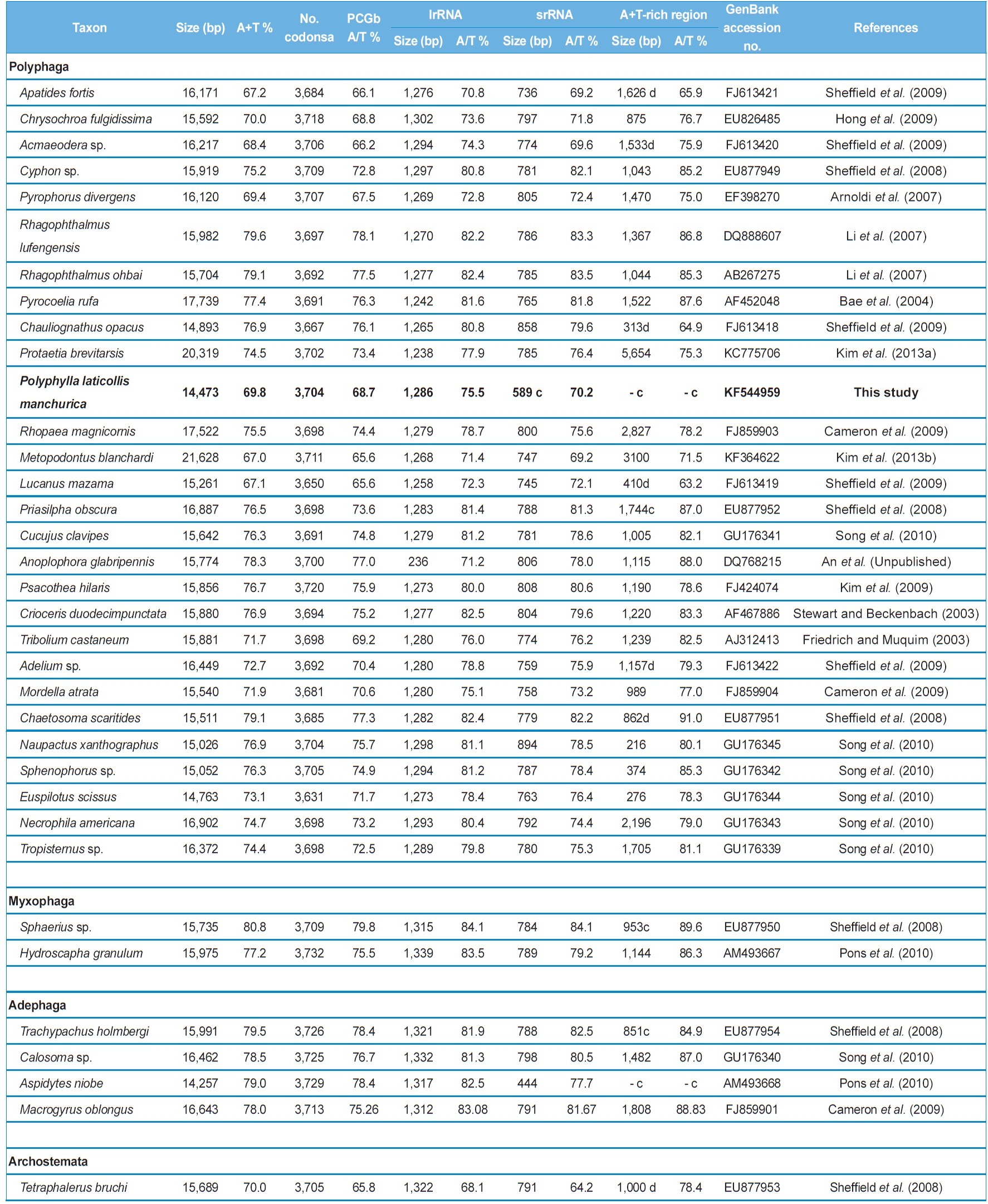
Characteristics of the coleopteran mitochondrial genomes sequenced in their entirety and near entirety
which contains a 4,051-bp long, large non-coding sequence located between tRNAIle and tRNAGln, along with a 3,100-bp long A+T-rich region, resulting in a genome size of 21,628 bp, which is at least 5 kb larger than typical animal mitogenomes (Kim
The
>
Overlapping and intergenic spacer sequences
The
Next longer intergenic spacer sequence is located between ND2 and tRNATrp as 14 bp (Table 2). A search on this region from other coleopteran insects has shown fluctuating length: only a few spacer sequences in most species (e.g., 4 bp in
The
All PCGs, with the exception of COI and ATPase8 of the
intergenic spacer sequences and gene overlaps. In this regard, they proposed asparagines (AAT or AAC) as the start codon for COI gene, because those are the first non-overlapping inframe codons, and found at the corresponding position in all sequenced Polyphaga in Coleoptera (Fig. 3; Sheffield
Eleven of the 13 PCGs have a complete termination codon of TAA or TAG, but the COII and COIII genes harbor the incomplete termination codon, T (Table 2). The most common interpretation of this phenomenon is that TAA termini are created via post-transcriptional polyadenylation (Ojala
The
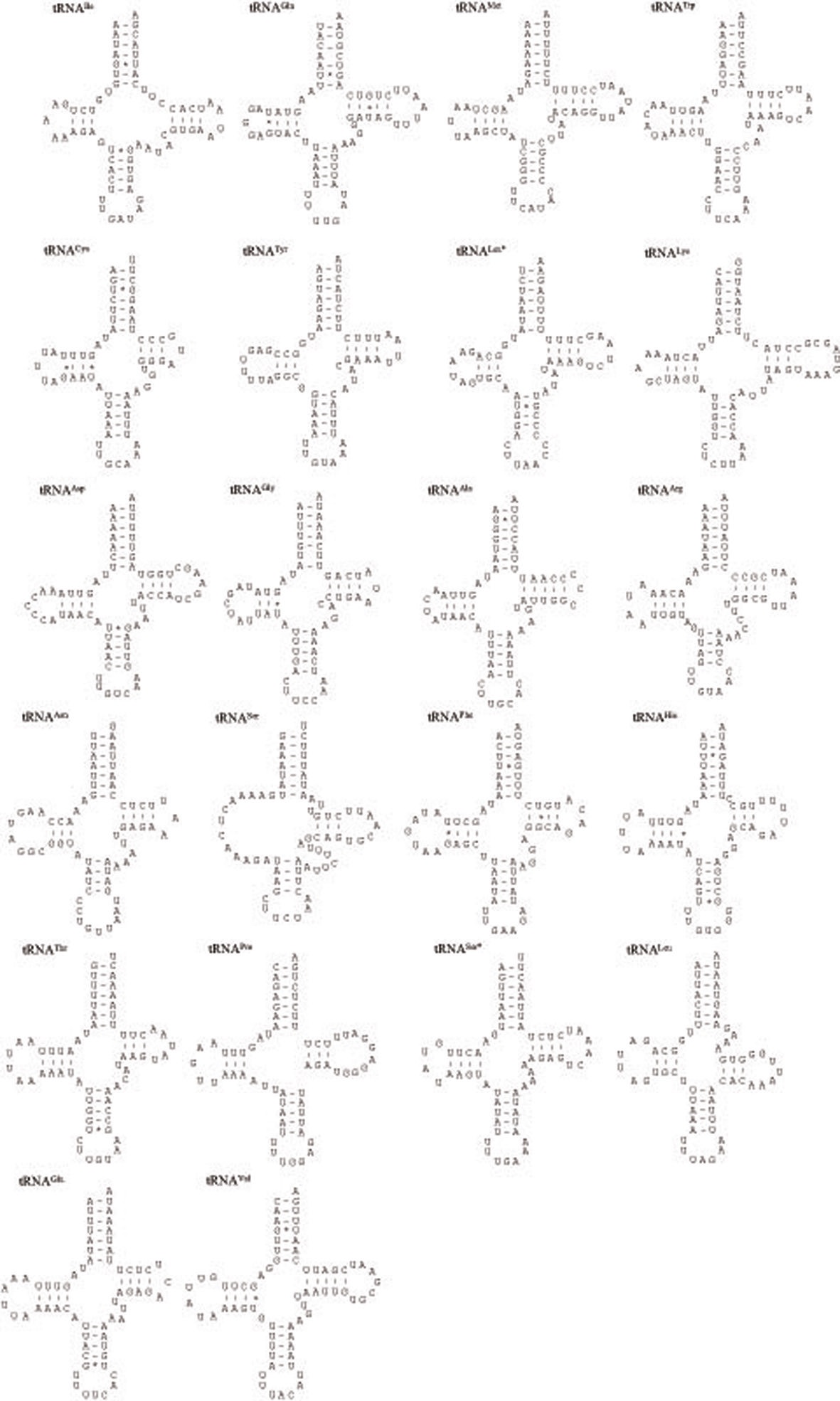
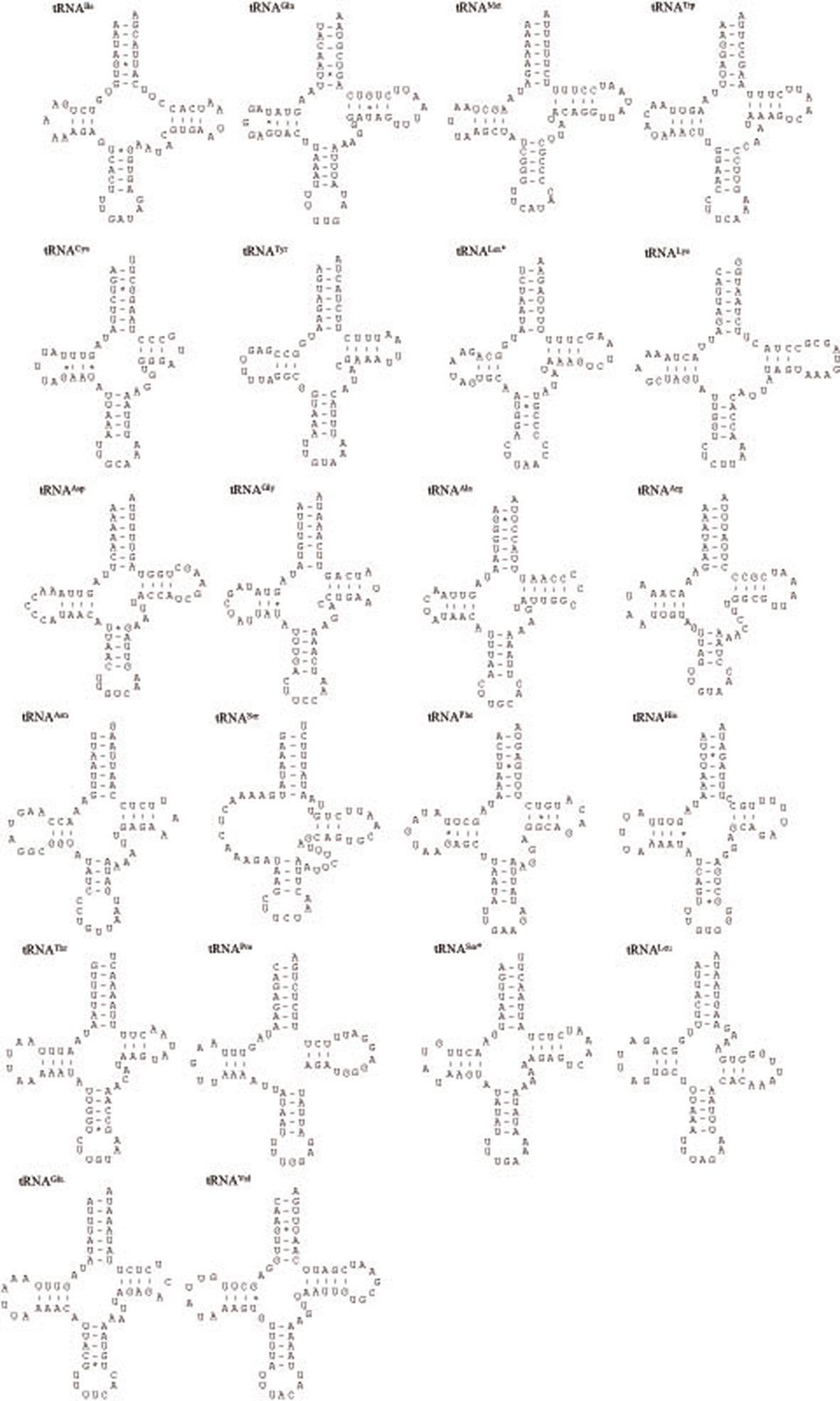
metazoan species, including insects (Wolstenholme 1992; Garey and Wolstenholme, 1989). For the proper function of a tRNA the DHU arm, which is involved in tertiary interaction requires proper folding (Rich and RajBhandary, 1976). The nuclear magnetic resonance analysis from nematodes has shown that the aberrant tRNASer(AGN) also functions in a similar way to that of usual tRNAs by structural adjustment to fit in the ribosome (Ohtsuki
The size of tRNAs ranged from 61 (tRNACys) to 71 bp (tRNALys) in
>
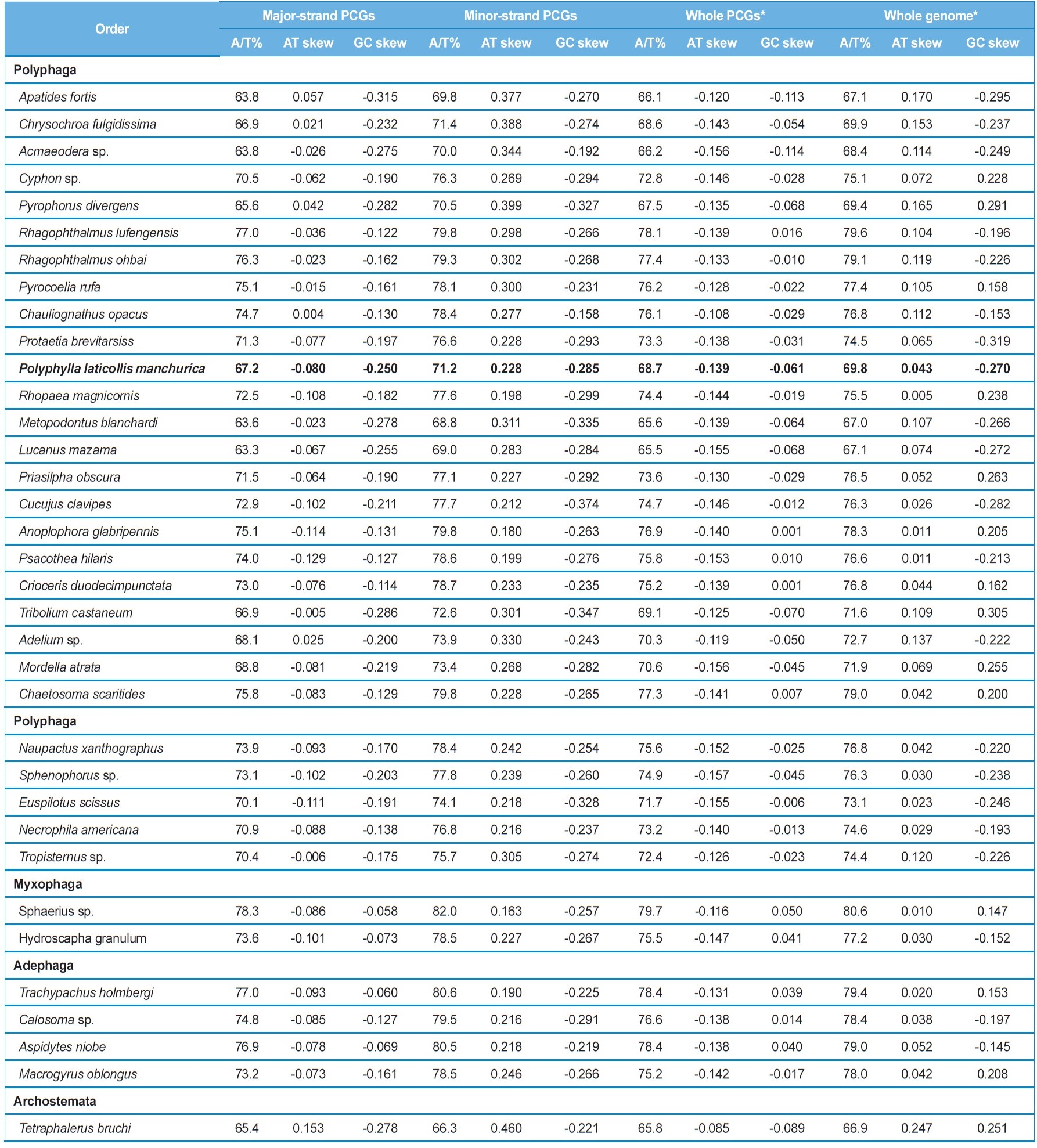
Nucleotide composition, codon usage, and skewness
The nucleotide composition of the mitogenome of
coleopteran insects, where these values range from 67.2% in
[Table 4.] Composition and skewness in the coleoptera mitochondrial genomes
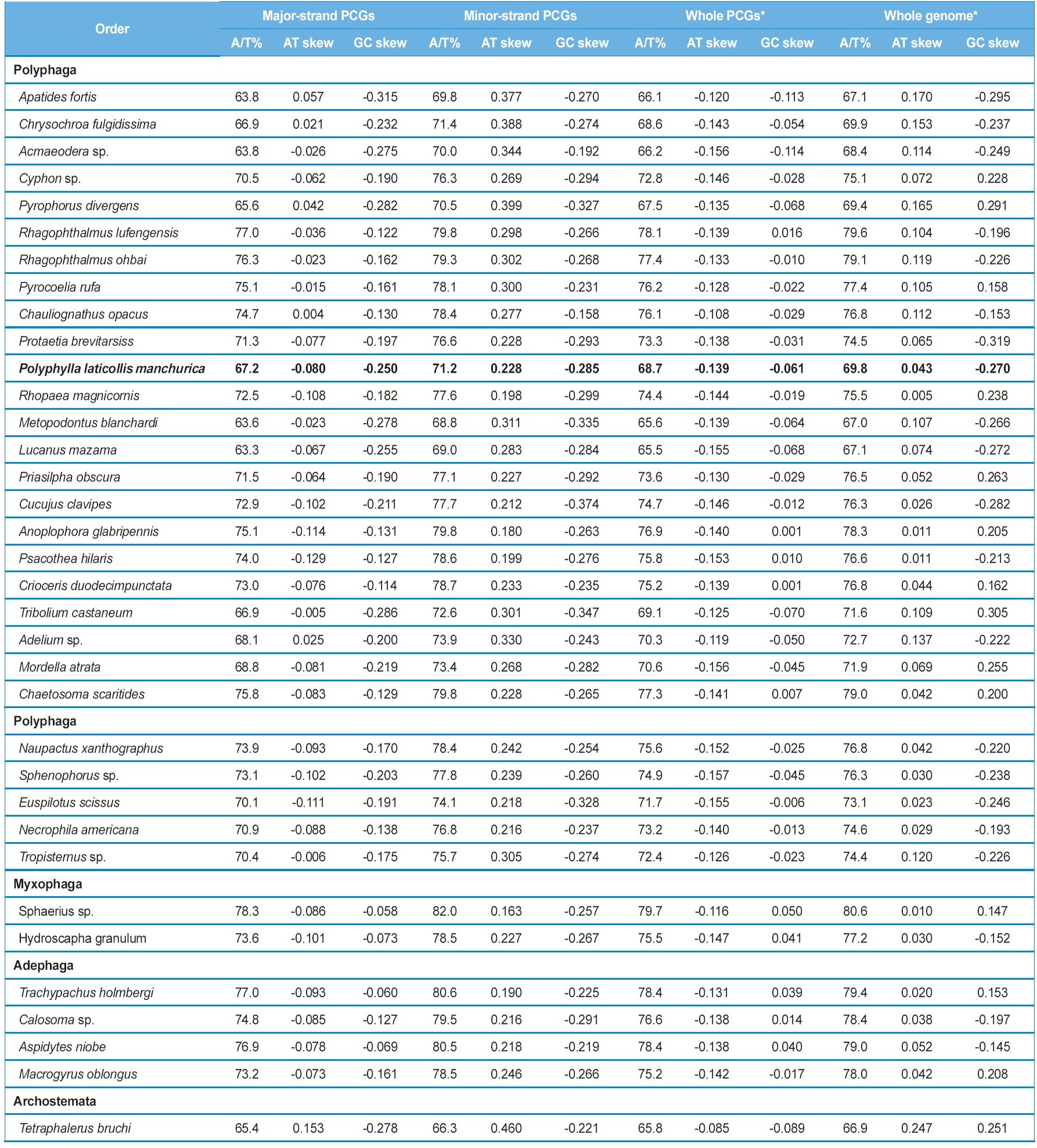
Composition and skewness in the coleoptera mitochondrial genomes
[Table 5.] Frequency of four most frequent codons of coleopteran insects

Frequency of four most frequent codons of coleopteran insects
ND3, ND6, COI, COII, COIII, ATPase6, ATPase8, and CytB) are encoded is slightly T-skewed (AT skew = -0.129 ~ 0.153), whereas the minor-strand, in which four PCGs (ND1, ND4, ND4L, and ND5) are encoded is obviously A-skewed (AT skew = 0.163 ~ 0.460), although both strands are C-skewed (Table 4). Thus, the two strands are sharply distinct in A/T-skewness, indicating that mutational pressures that favor Ts or As are starkly different between the two strands. It has been suggested that the lagging strand, equivalent to the major strand, should be more prone to chemical conversion of As to Gs and Cs to Ts, by the mechanism called deamination than the leading strand, and this may have resulted in the enriched Ts and Gs in the lagging strand, and As and Cs in the leading strand (Reyes
The genome-wise A/T bias is also reflected in the codon usage of
The analysis of the base composition at each codon position of the concatenated 13 PCGs of

Summary of base composition at each codon position of the concatenated 13 PCGs in coleopteran mitochondrial genomes