Cells derived from multicellular organisms provide a more convenient and detailed analytic system than those using whole organisms. Establishing permanent cell lines
Here, we have focused on marine medaka,
Marine medaka
Fish serum was isolated from Nile tilapia
>
Preparation of embryo extract
For the preparation of embryo extract from
>
Isolation and culture of blastomeres from blastula embryos
Embryos (8-20) developed to the early or late blastula stage were rinsed with 70% ethanol for 10 s to prevent microbial contamination, and washed three times in Dulbecco’s phosphate-buffered saline (DPBS; Gibco, Grand Island, NY, USA). The developmental stage of embryos was identified using the method of Song et al. (2009). Briefly, the early blastula stage occurs 7 h 45 min-9 h after fertilization, while the late blastula stage occurs 9 h-11 h 30 min after fertilization. The embryos were subsequently ruptured by fine needles in a petri dish and the scattered blastomeres were transferred to a centrifuge tube after removing the eggshells. After centrifugation, isolated blastomeres were cultured on 0.1% gelatin (Sigma-Aldrich, St. Louis, MO, USA)-coated tissue culture plates (48 multi-well plate; SPL Life Sciences, Pocheon, Korea) with Dulbecco’s Modified Eagle’s Medium (Gibco) supplemented with 4.5 g/L D-glucose, 20 mM HEPES, 1% (v/v) non-essential amino acids (Gibco), 15% (v/v) fetal bovine serum (FBS; Cellgro, Manassas, VA, USA), 1% (v/v) fish serum, embryo extract (50, 100, or 150 μg/mL), 1% (v/v) penicillin-streptomycin mixture (Gibco), 10 ng/mL recombinant human basic fibroblast growth factor (bFGF; Gibco), 100 μM β-mercaptoethanol (Gibco), 2 nM sodium selenite (Sigma-Aldrich), and 1 mM sodium pyruvate (Gibco). The embryo extract concentration was fixed at 50 μg/mL after seven passages. All cell culture was conducted using the culture medium above, unless stated otherwise. The cells were cultured in a 28℃ incubator with an air atmosphere and sub-cultured every 2 or 3 days when they reached 80-90% confluency.
To determine growth rates, 2 × 104 cells per well were seeded in 0.1% gelatin-coated 48-well culture plates filled with culture medium and cultured for 10 days in a 28℃ incubator with air atmosphere. Culture medium was changed every 3 days after first change at day 1 of culture. From day 1 to 10 of culture, proliferating cells were counted daily with a hemocytometer (Marienfeld, Lauda-Konigshofen, Germany) after detachment by 0.05% trypsin-EDTA (Gibco). This experiment was repeated three times in a independent manner.
The cells were washed in DPBS and treated with a 0.075 M KCl (Sigma-Aldrich) solution for 10 min at 28℃. The swollen cells were fixed with a cold fixative solution of 3:1 methanol (Sigma-Aldrich) and acetic acid (Sigma-Aldrich) changed three times by centrifugation at 400
A total of 3,600 cells were initially seeded on 60-mm tissue culture plates (SPL Life Sciences) in culture medium for 10 days under an air atmosphere at 28℃. The medium was replaced every 3 days and clonally expanded colonies counted at the end of culture.
>
Growth rates according to medium composition
To evaluate the growth reactivity of cells to culture medium supplements, 1,000 cells/well were cultured in a 0.1% gelatincoated 96-well culture plate with culture medium supplemented with various concentrations of fish serum (0, 0.5, 1, and 1.5%), FBS (0, 5, 10, 15, and 20%), bFGF (0, 5, 10, and 15 ng/ mL), or embryo extract (0, 25, 50, 75, and 100 μg/mL). After 6 days of culture, the viability of cultured cells was assayed using the cell counting kit-8 (Dojindo, Kushu, Japan), according to the manufacturer’s instructions.
The Statistical Analysis System (SAS) software was used to analyze the numerical data. When an analysis of variance (ANOVA) identified a significant primary effect, treatments were subsequently compared by the least-square or Duncan’s method.
>
Establishment of permanent cell lines
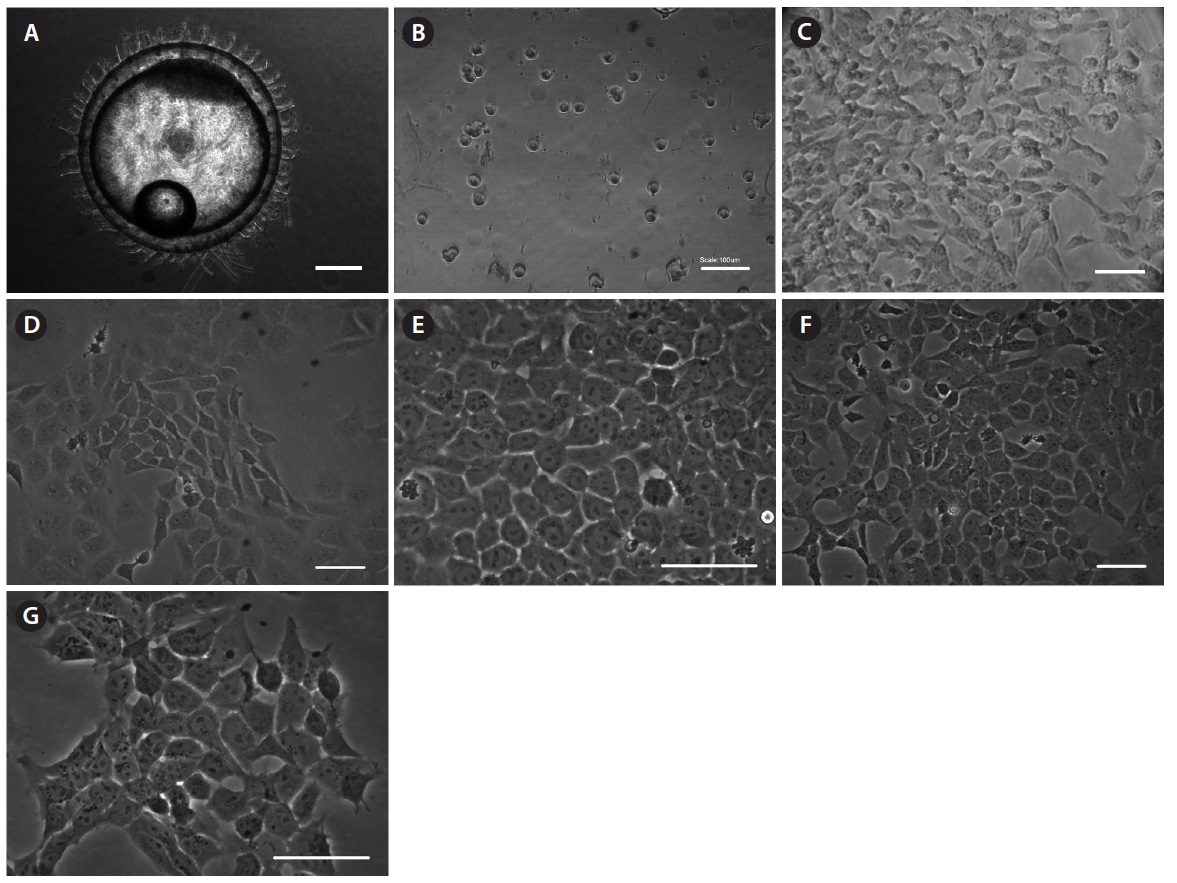
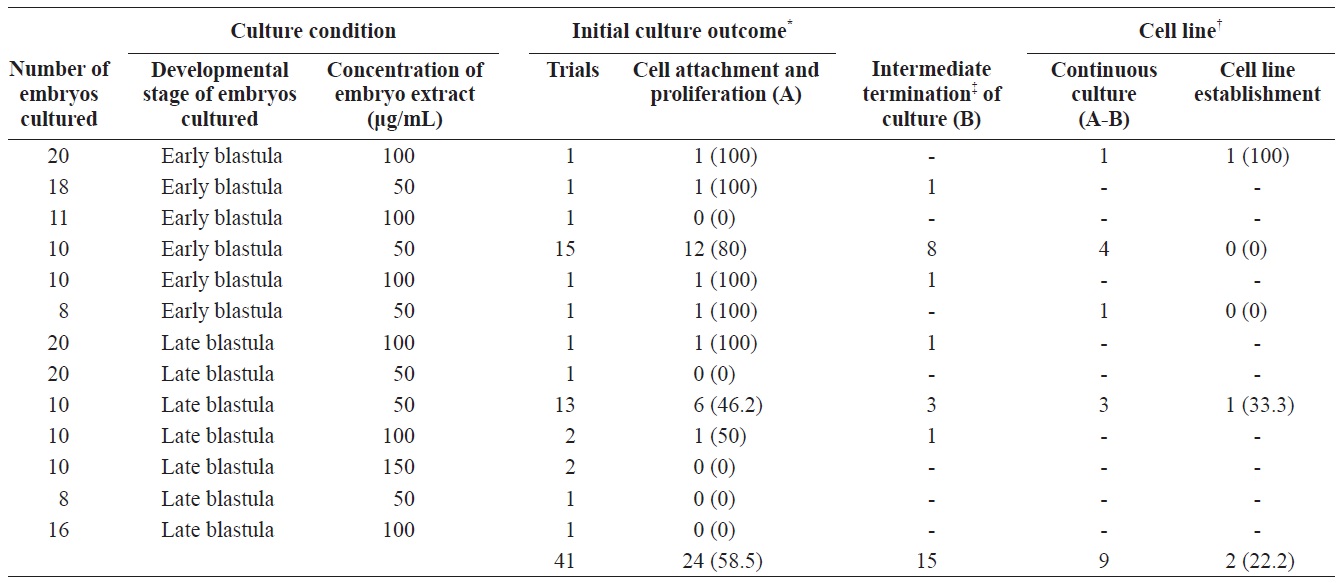
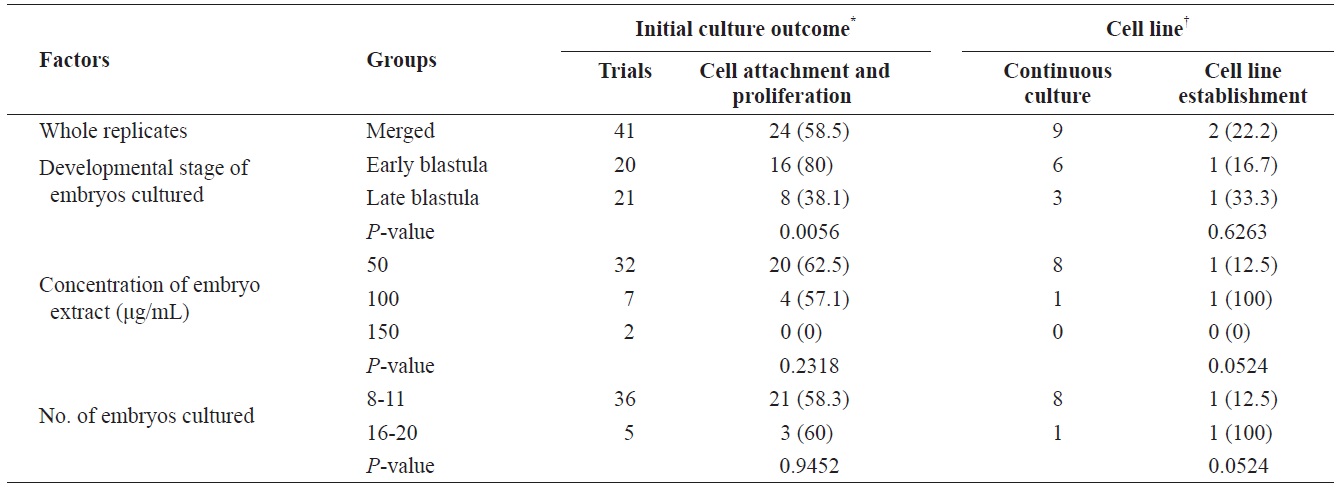
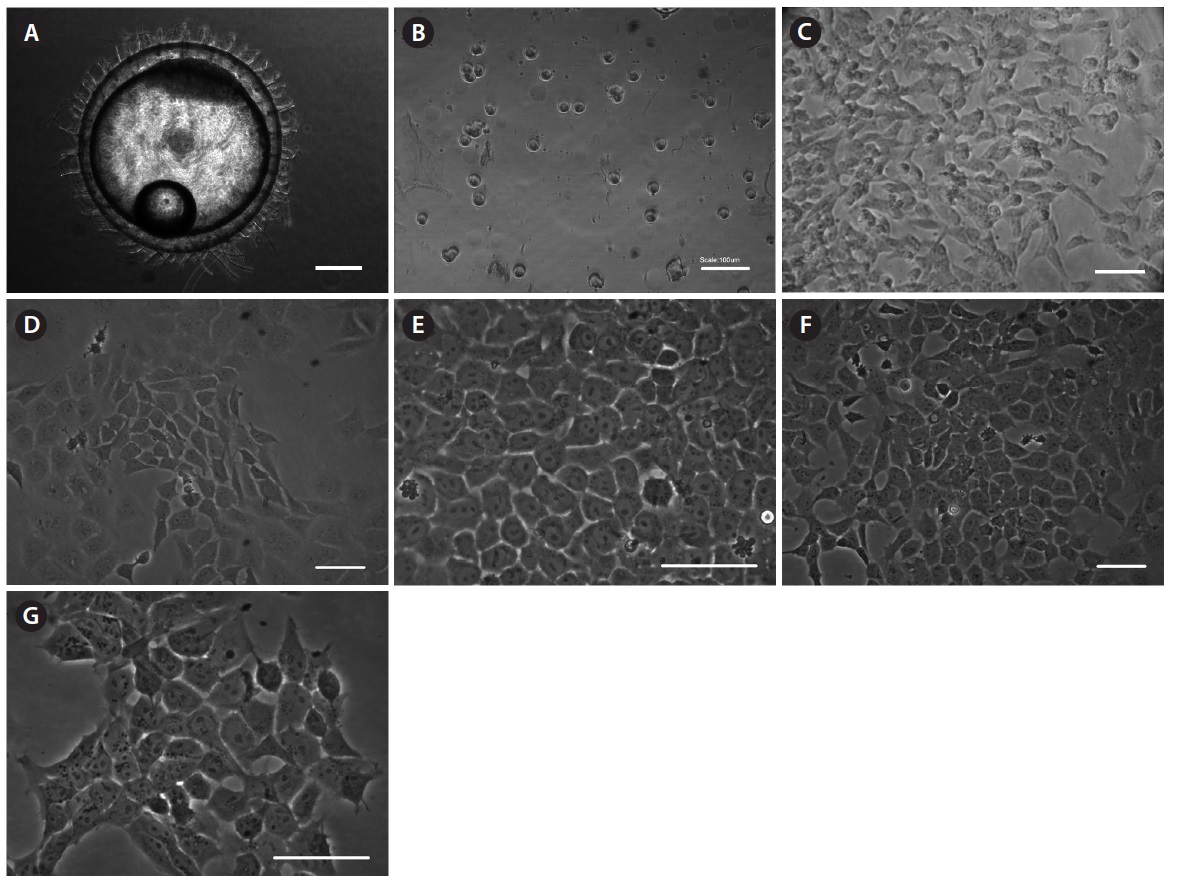
To establish permanent cell lines, we isolated blastomeres from developing embryos (Fig 1A and 1B). Isolated blastomeres from early or late blastula of a number of embryos were cultured under different conditions. As shown in Table 1, in 24 (58.5%) of 41 trials, the formation of primary cells was observed from cultured embryonic cells, and these cells proliferated sufficiently for subculture after 7 days (Fig. 1C). Nine of these trials were continuously cultured without intermediate termination, while all but two cases failed to induce immortalization. We found that the developmental stage of embryos was a key factor contributing to primary cell attachment and proliferation during initial culture (Table 2). A higher frequency of success was detected when blastomeres from early blastulas were used, compared to late blastulas (80 vs. 38.1%,
>
Growth properties of permanent cell lines
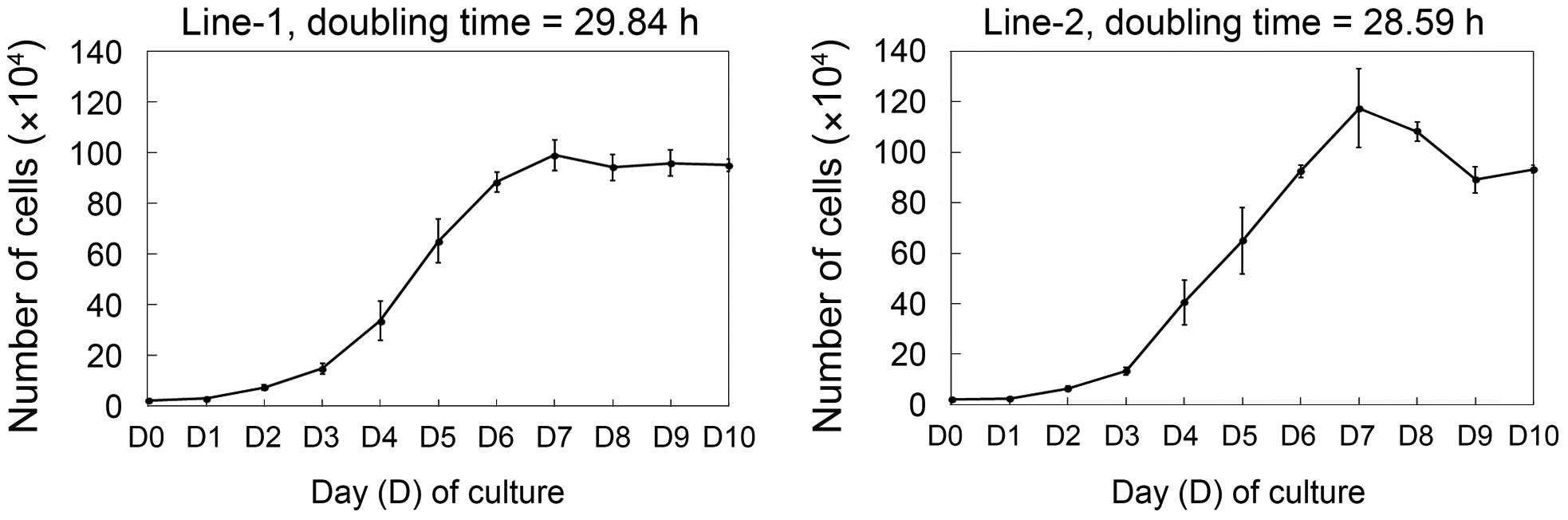
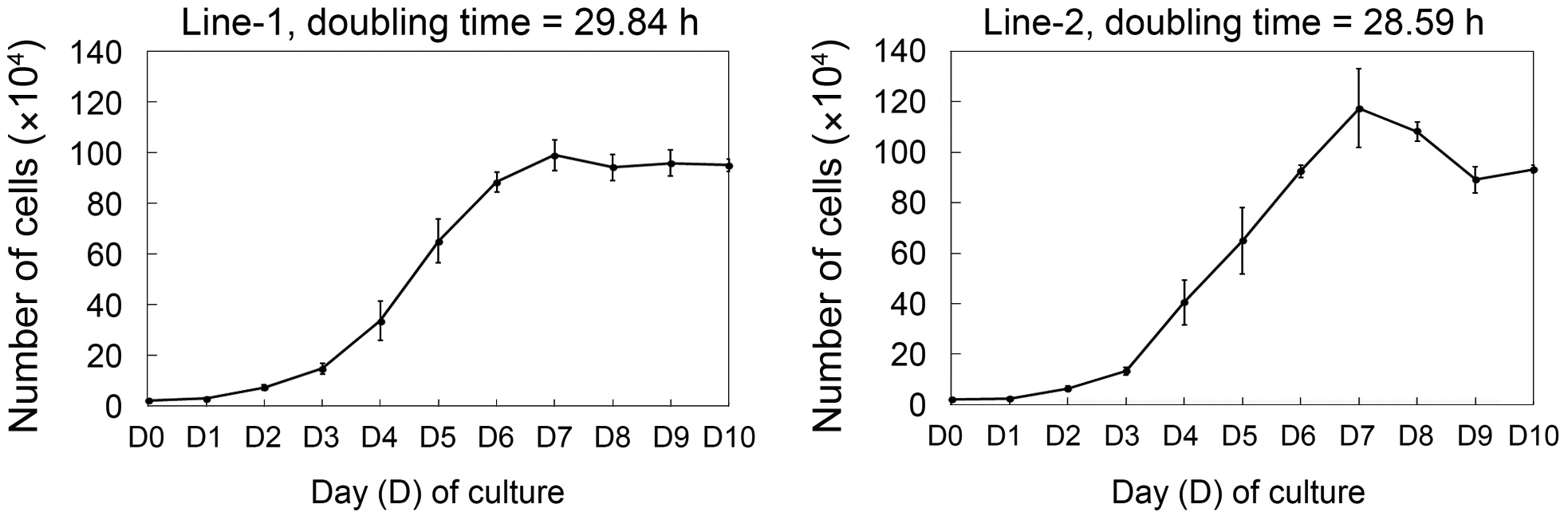
The growth curves of established cell lines are shown in Fig. 2. Cell line 1 had a doubling time of 29.84 h and cell
[Table 1.] Culture outcome of embryonic cells from blastula stage embryos
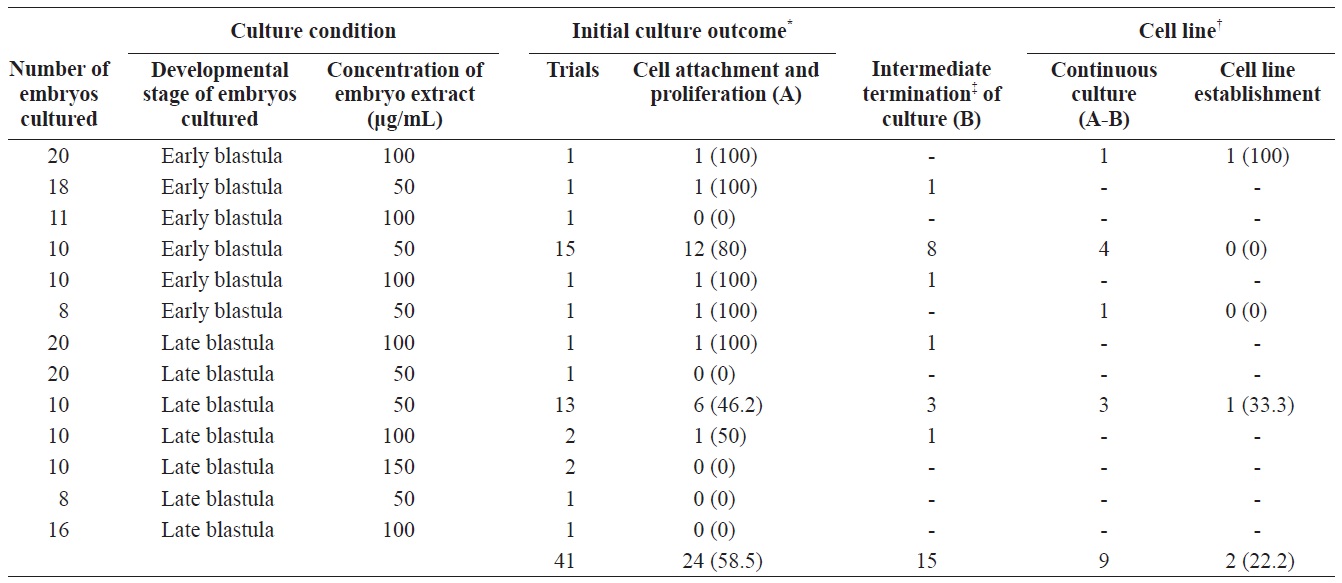
Culture outcome of embryonic cells from blastula stage embryos
[Table 2.] Factors affecting initial culture outcome of embryonic cells
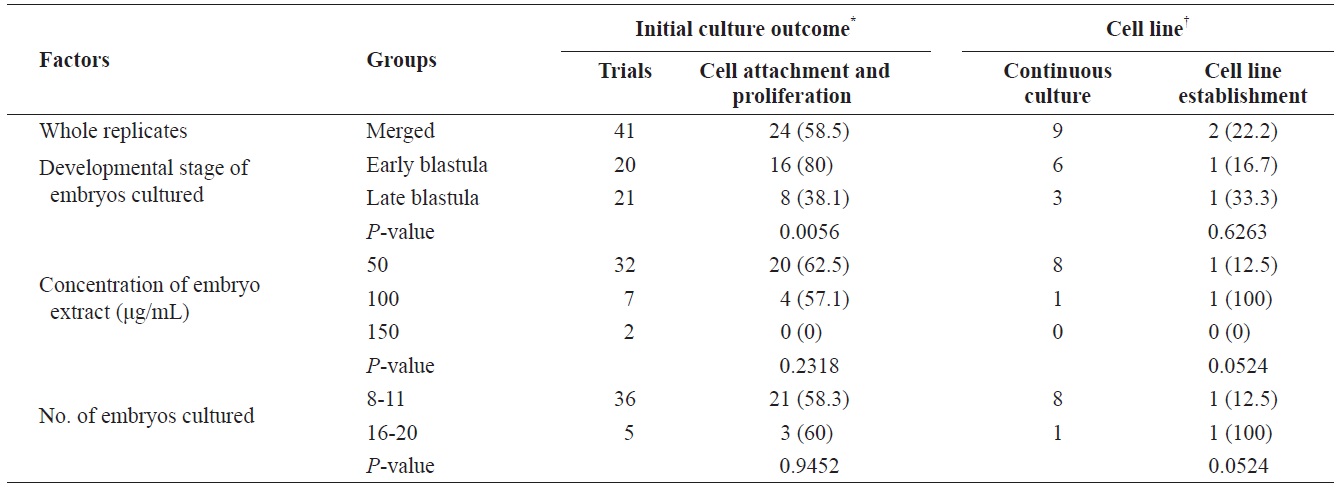
Factors affecting initial culture outcome of embryonic cells
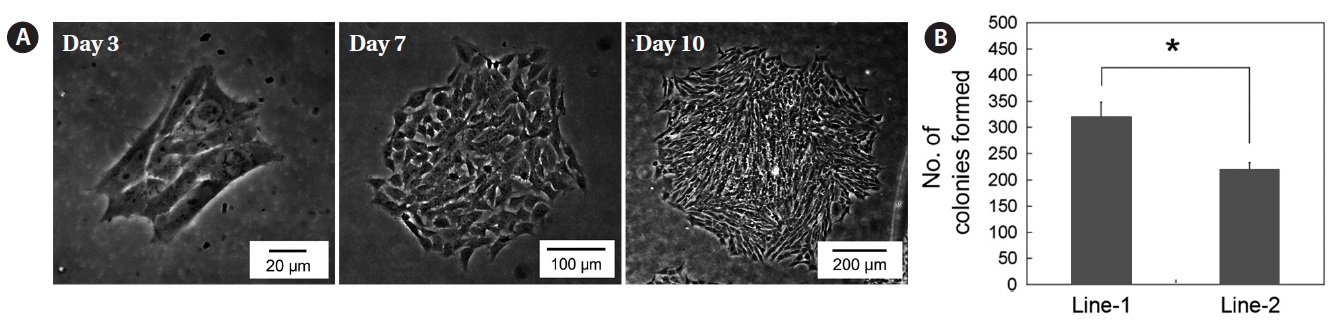
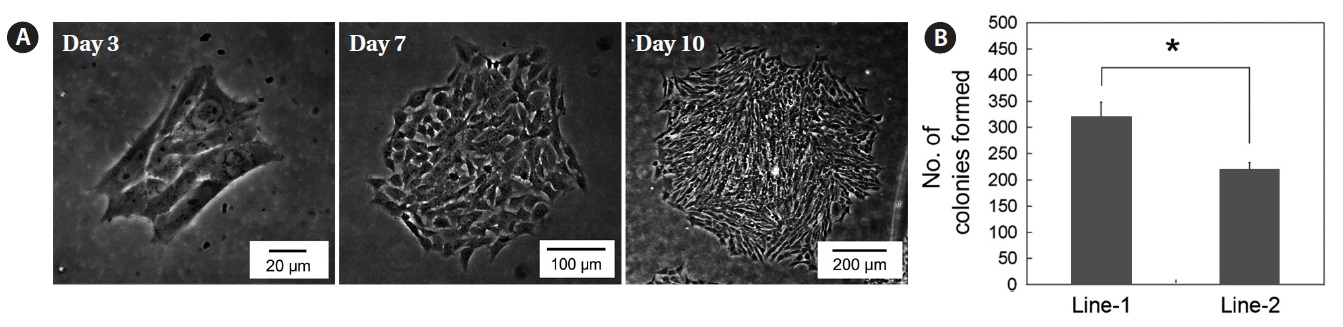
line 2 had a doubling time of 28.59 h. To determine whether these cell lines could expand clonally, the cells were cultured at a low density (127 cells/cm2) and colonies formed by clonal expansion were identified and counted. As shown in Fig. 3A, small colonies derived from single cells were identified on day 3 of culture and these colonies grew continuously. The total number of colonies on day 10 of culture was significantly different between the two cell lines―clonogenicity was higher in cell line 1 than cell line 2 (327 ± 27.8 vs. 220 ± 12.2,
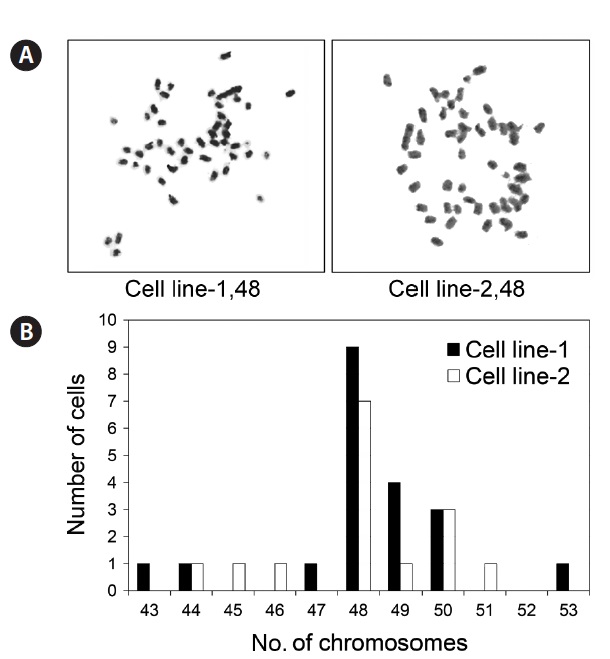
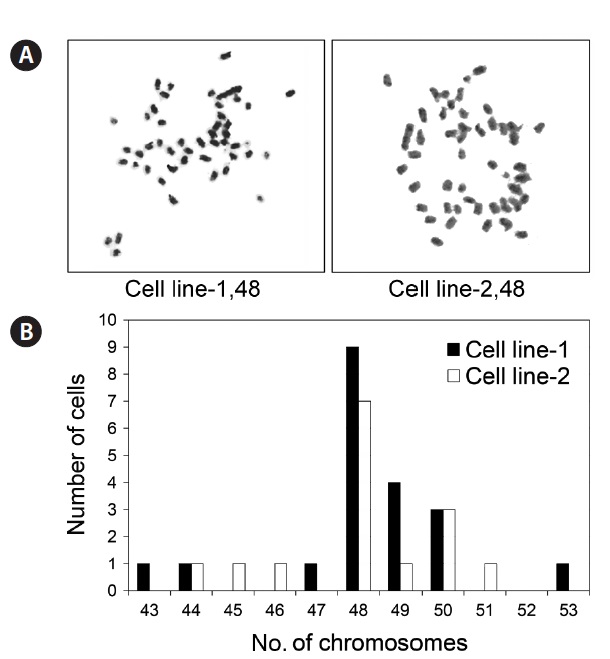
To evaluate the chromosomal normality of the established cell lines, metaphase chromosomes were prepared and the chromosomes in each cell counted. As shown in Fig. 4, a proportion of cell population bearing the normal number of 48 chromosomes was 45% and 46.7% in cell line 1 and 2, respectively.
>
Growth responses to different media supplements
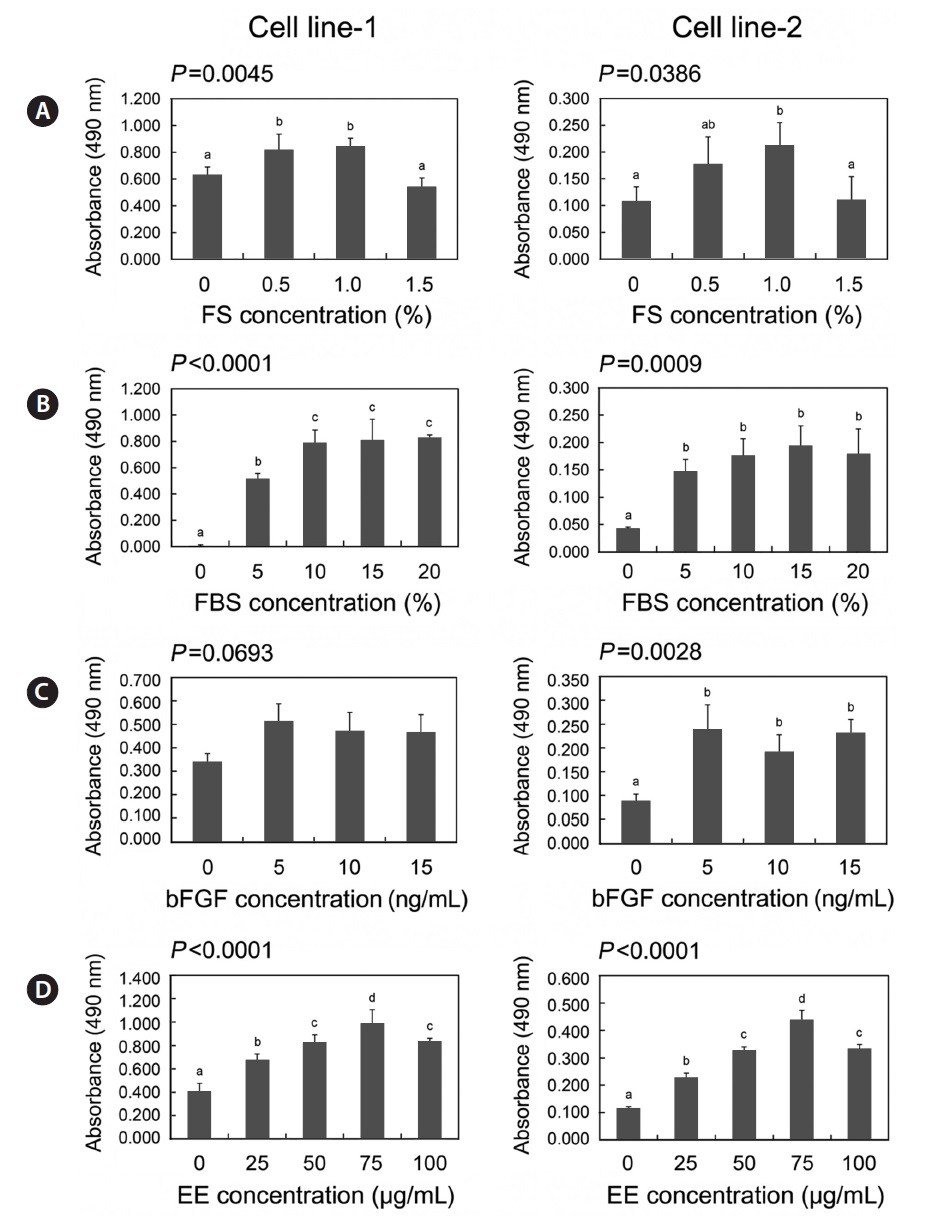
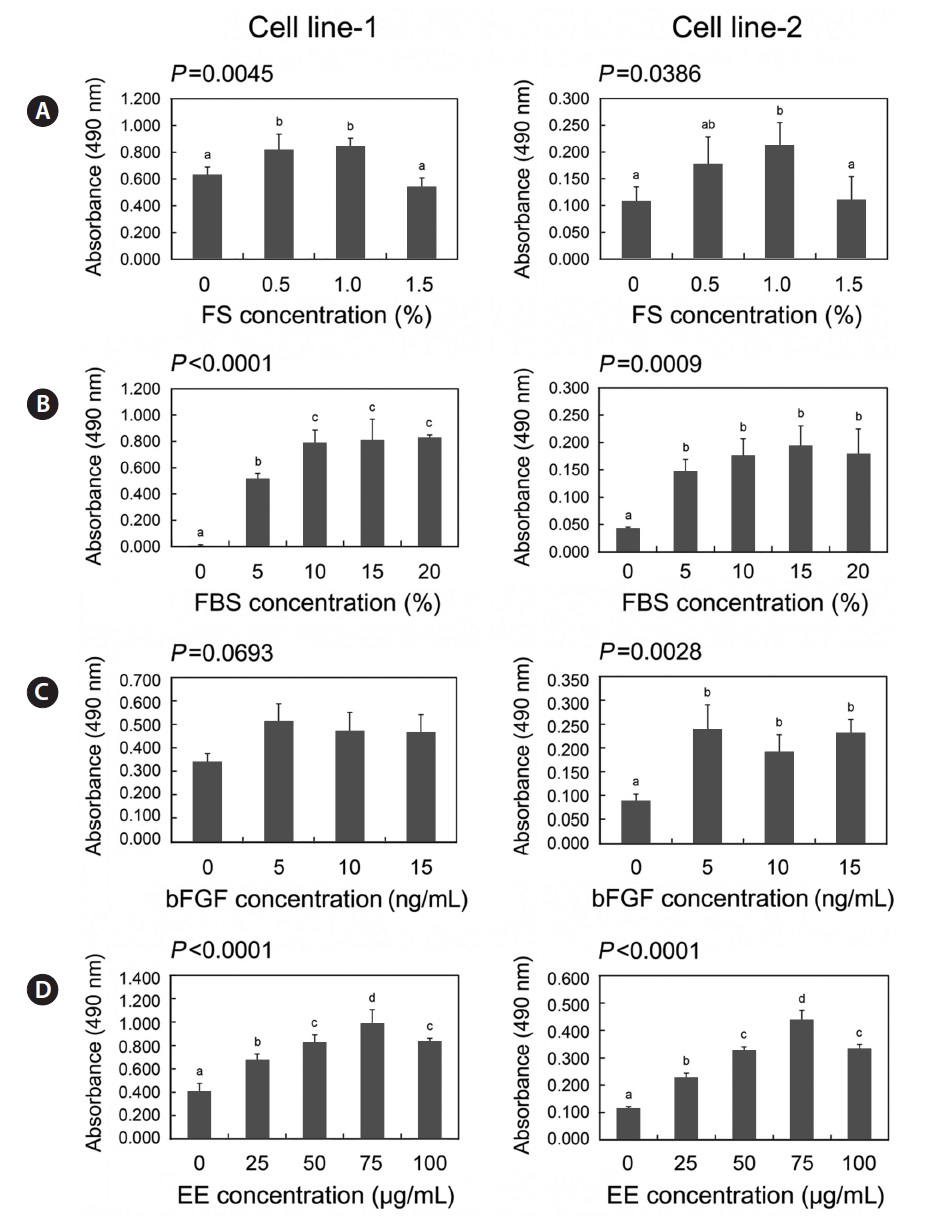
As a part of optimization of culture conditions, we assessed the growth response to the following medium supplements: fish serum, FBS, bFGF, and embryo extracts. As shown in Fig. 5, removal of each supplement from the medium resulted in significant inhibition of the growth of both cell lines (
>5 ng/mL bFGF (Fig. 5C). Embryo extracts stimulated growth of both cell lines in a dose-dependent manner; however 100 μg/mL inhibited the growth of both cell lines (Fig. 5D).
We have shown that cell lines from marine medaka
To establish permanent cell lines from embryonic cells at the blastula stage, the number and developmental stage of the embryos, as well as the embryo extract concentration, were varied and cell attachment, proliferation, and cell line establishment subsequently evaluated. We found that the developmental stage of embryos, as either early or late blastulas, was a key factor in the attachment and proliferation of cultured embryonic cells. This may a consequence of the midblastula transition, which results in dramatic changes in embryonic cells, such as cell cycle elongation, desynchronization of cells, zygotic transcription, and commencement of cell motility (Newport and Kirschner, 1982; Kane and Kimmel, 1993, Dalle Nogare et al., 2009). Early blastula cells exhibit rapid proliferation rate due to repeating S and M phases without a gap period, which may aid in attachment and proliferation in extraordinary environments. The number of embryos in initial culture is another important factor that affects cell density, which plays a critical role in the regulation of the function and differentiation of many cell types (Limoli et al., 2004; Portela et al., 2010; Yi et al., 2010). Based on our results, more than 10 embryos/well of a 48-well plate provided sufficient primary cells to establish permanent cell lines from embryos. Embryo extracts are regarded as a key factor regulating the cell cycle and cell potency; indeed, 50 μg/mL was sufficient for initial cell culture and maintenance. Our data suggested that the event of cellular transformation of cultured embryonic cells into immortal state occurred at a frequency of 22.2% (two lines from nine primary cultured cells) in a spontaneous manner. However, it is unknown whether the immortalization rate involved these factors due to a lack of replicates; therefore, further studies are necessary.
The ability of a single cell to grow to form a colony is a phenotypic marker of transformed cells, which possess the capacity for unlimited division and lose density limitation of proliferation (Franken et al., 2006; Freshney, 2010). In this study, 9% of the cells from cell line 1 and 6% from cell line 2 possessed clonogenicity, confirming that the two established cell lines underwent successful spontaneous transformation to be permanent cell lines. In addition to the phenotypic changes, we identified a high frequency of chromosomal abnormality (53.3-55%) in the established cell lines, which has been associated with cellular transformation (Rao et al., 2006; Thibodeaux et al., 2009; Stepanenko and Kavsan, 2012). Genetic mutations in cell cycle, survival, or apoptosis regulatory genes under suboptimal culture conditions can induce chromosomal instability and cellular transformation (Li et al., 2010; Thompson and Compton, 2010; Weiss et al., 2010). Nevertheless, the selection of cells with chromosomal normality and subsequent optimization of culture conditions enables use of a normal cell population, if required.
For the culture of established cell lines, culture medium was supplemented with the following four components: fish serum, FBS, bFGF, and embryo extracts, which promote proliferation (Collodi and Barnes, 1990; Boilly et al., 2000; Freshney, 2010). Knowledge of the dependence of the cell lines on these factors makes it possible to evaluate cellular properties and optimize culture conditions. The growth of the two established cell lines was influenced by all four factors, with the exception of bFGF. This suggests that, in spite of the acquisition of the ability to grow continuously, these two cell lines retained normal processes in terms of handling signals from exogenous growth factors, which can be destroyed by cellular transformation (Eagle et al., 1970; Li et al., 1989). We also found that the two established cell lines had different growth performances in this experiment, despite similar responses to the medium supplements. Such differences are not matched with the result that showed similar growth rates between two cell lines, which may have been due to differences in the cell density used between two experimental sets. The initial cell density for the growth rate experiments was 31,454 cells/cm2 while it was 2,600 cells/cm2 for the experiments of growth responses to main media supplements. This suggests that cell line 2 was more susceptible to culture environments at a low cell density than cell line 1, which is supported by the significantly lower clonogenicity of cell line 2 compared to cell line 1.
We targeted blastomeres from blastula-stage embryos to generate permanent cell lines in this fish species. Blastomeres at this stage have the potential to develop into many types of cells and tissues (Nilsson and Cloud, 1992; Hong et al., 1998). Thus, cell lines established from blastomeres at the blastula stage might have a developmental potential similar to that of the original blastomeres. The establishment of such cell lines, called embryonic stem (ES) cells or ES-like cells, has been reported (Hong et al., 1996; Bejar et al., 2002; Yi et al., 2009; Hong et al., 2011). These cell types are good models for investigation of cellular and genetic events during
In conclusion, we have described the basic characteristics of two permanent cell lines derived from the embryos of