D-glucose-6-phosphate (G6P), glucose-6-phosphate dehydrogenase (G6PDH), honokiol, magnolol, β-nicotinamide adenine dinucleotide phosphate (β-NADP+), magnesium chloride (MgCl2), potassium phosphate (KH2PO4), and terfenadine were purchased from Sigma-Aldrich (St. Louis, MO, USA). Solvents were LC-MS grade (Fisher Scientific Co., Pittsburgh, PA, USA) and the other chemicals were of the highest quality available. Pooled human liver microsomes (coded HLM 150) were obtained from BD Biosciences (Woburn, MA, USA). The manufacturer supplied information regarding protein content, P450 content, and enzyme activity (available: www.bdgiosciences.com).
>
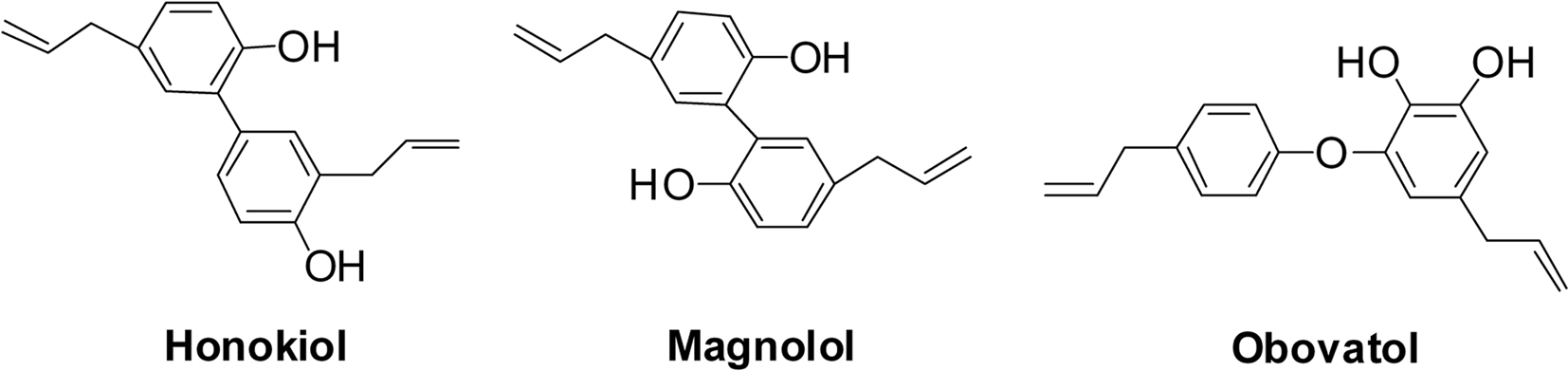
Inhibitory effects of honokiol and magnolol on P450 activity
The inhibitory potency of honokiol and magnolol were determined with cytochrome P450 assays in the absence and presence of these compounds (final concentration of 0.5~ 50 μM with methanol concentration less than 0.5%) using pooled human liver microsomes. Phenacetin
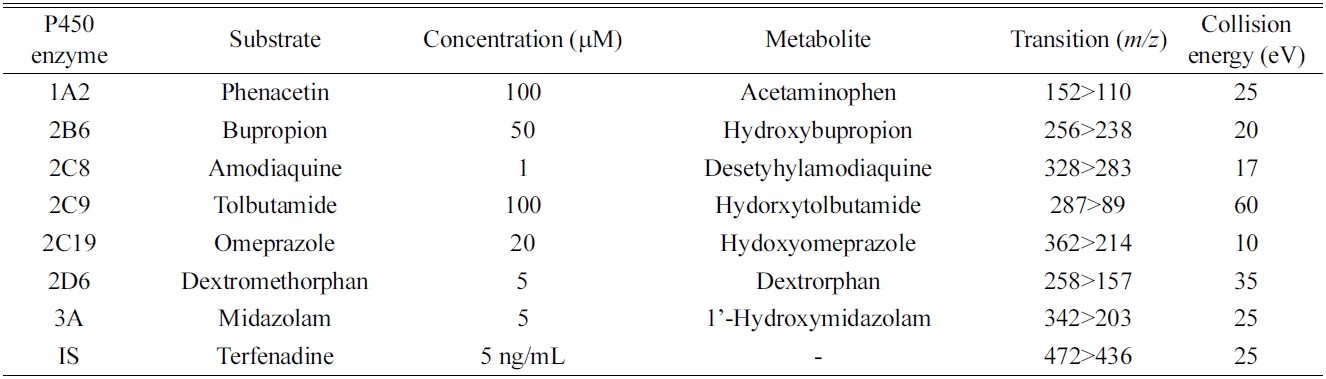
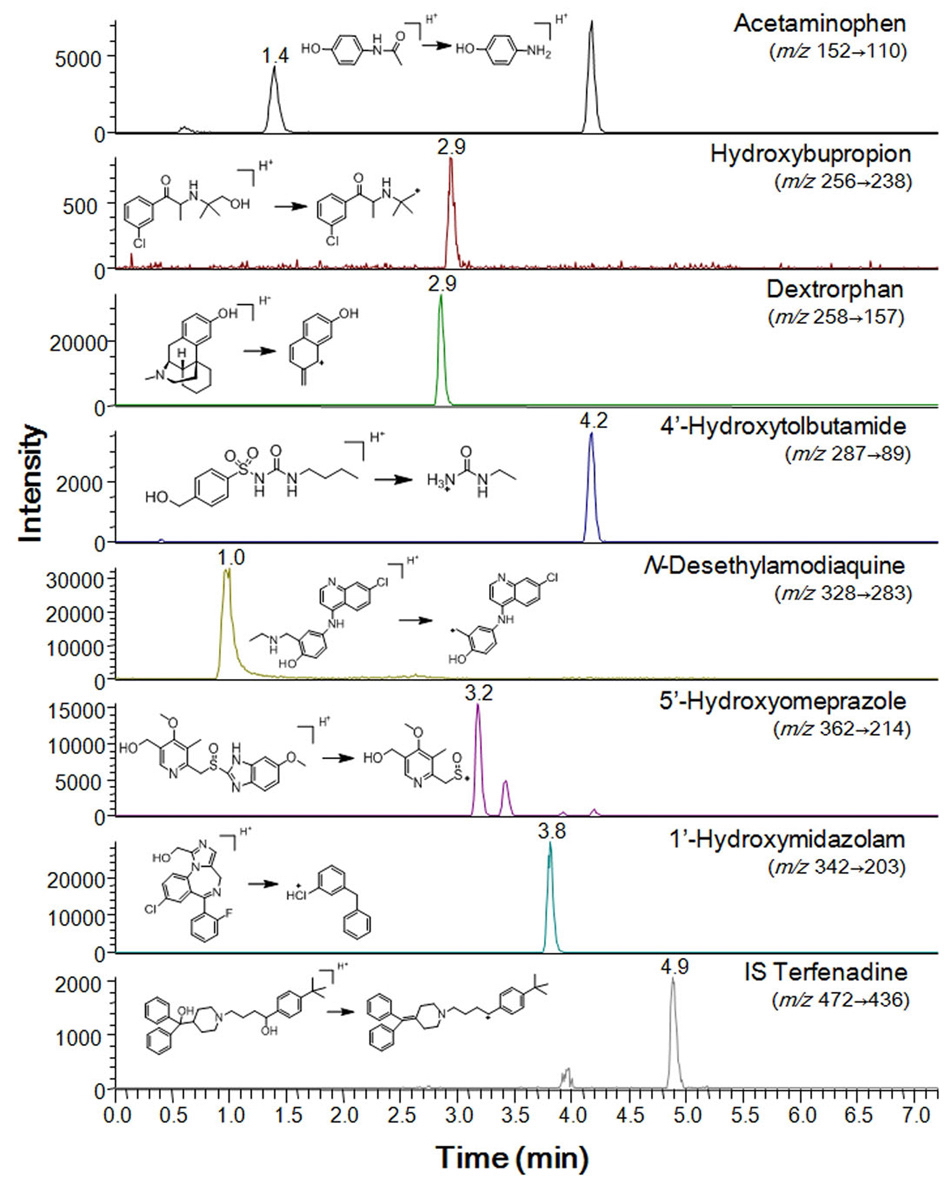
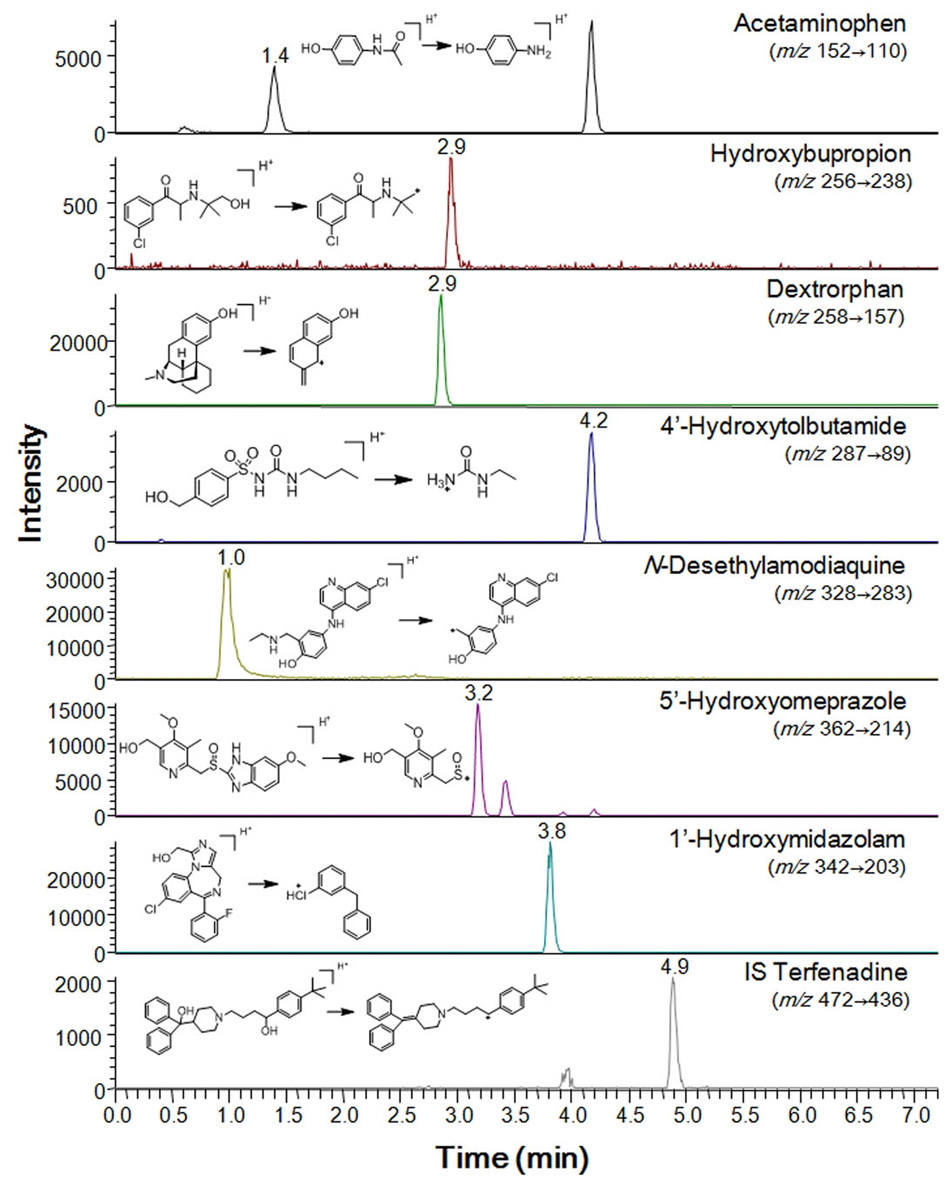
All metabolites of the P450 isoform-specific substrates were measured by tandem mass spectrometry as described previously.7,8 Briefly, the system consisted of a Thermo Vantage Triple quadrupole Mass Spectrometer (Thermo Fischer Scientific, San Jose, CA, USA), coupled with an Thermo Accela HPLC system (Thermo Fischer Scientific). The separation was performed on a Luna C18 column (2 mm i.d. × 30 mm, 5 μm, 100 A, Phenomenex, Torrance, CA, USA). The mobile phase consisted of 0.1% formic acid in acetonitrile (A) and 0.1% formic acid in water (B). Gradient elution was conducted as follows: 8% A was linearly increased to 95% over 7 min, and then immediately stepped back down to 8 % for re-equilibration until 11 min, at a flow rate of 0.2 mL/min. The electrospray interface was operated in positive ion mode at 4000 V. The operating conditions were as follows: capillary temperature, 350℃; vaporizer temperature, 300℃; sheath gas pressure, 35 Arb; Auxiliary gas, 10 Arb; nitrogen gas flow rate, 8 L/min. Quantitation was performed by selected reaction monitoring (SRM) of the [M+H]+ ion and the related product ion for each metabolite. The SRM transitions and collision energies determined for each metabolite and internal standard are listed in Table 1. The analytical data were processed by Xcalibur (version 2.1).
The enzyme inhibition parameter IC50 values (concentration of the inhibitor causing 50% inhibition of the original enzyme activity) were calculated using nonlinear leastsqua- res regression analysis of the plot of percent control activity versus concentration of honokiol or magnolol, using Win-Nonlin version 4.0 (Pharsight, Mountain View, CA, USA).
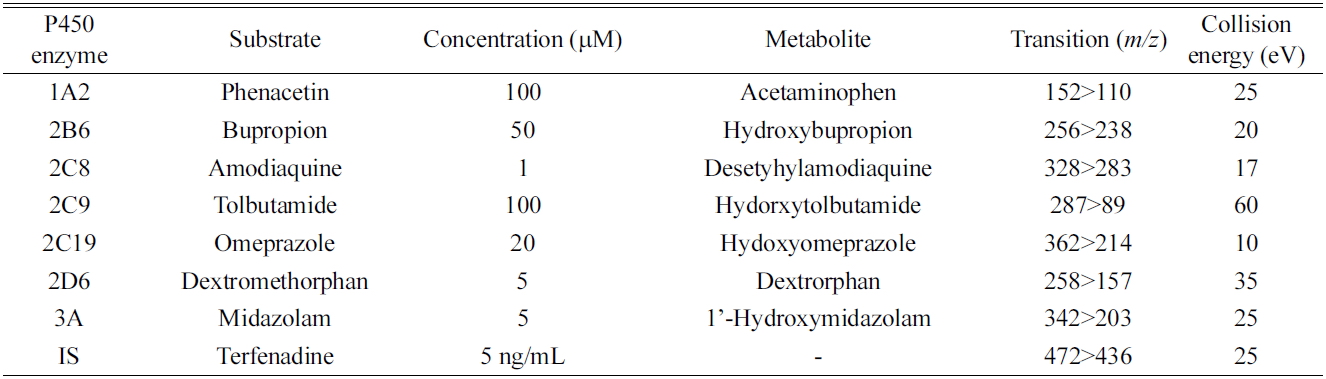
SRM parameters for the major metabolites of the seven P450 probe substrates used in all assays
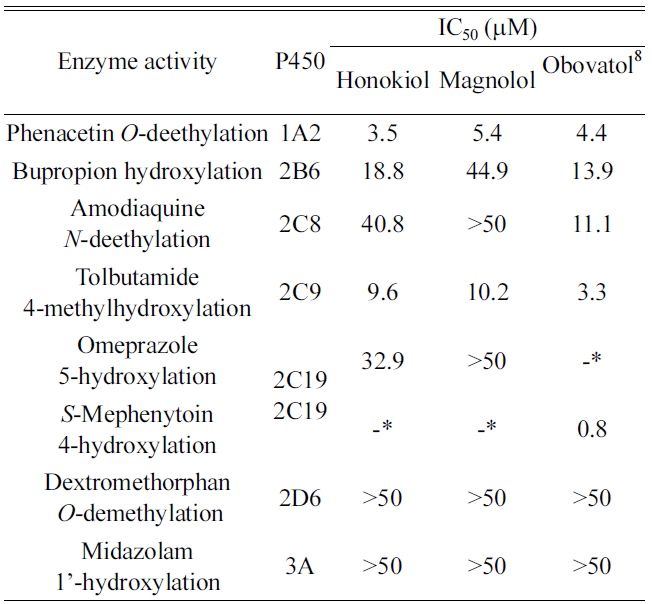
Inhibition of P450 activity was evaluated at concentrations of up to 50 μM honokiol and magnolol to investigate their effects on P450-mediated drug interactions in human liver microsomes. The activities of CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A were determined in human liver microsomes using a cocktail assay containing phenacetin for CYP1A2, bupropion for CYP2B6, amodiaquine for CYP2C8, tolbutamide for CYP2C9, omeprazole for CYP2C19, dextromethorphan for CYP2D6, and midazolam for CYP3A. The LC-MS/MS system in SRM mode was optimized for detection of each metabolite (Table 1 and Figure 1). Honokiol and magnolol inhibited the metabolism of CYP1A2-mediated phenacetin deethylase
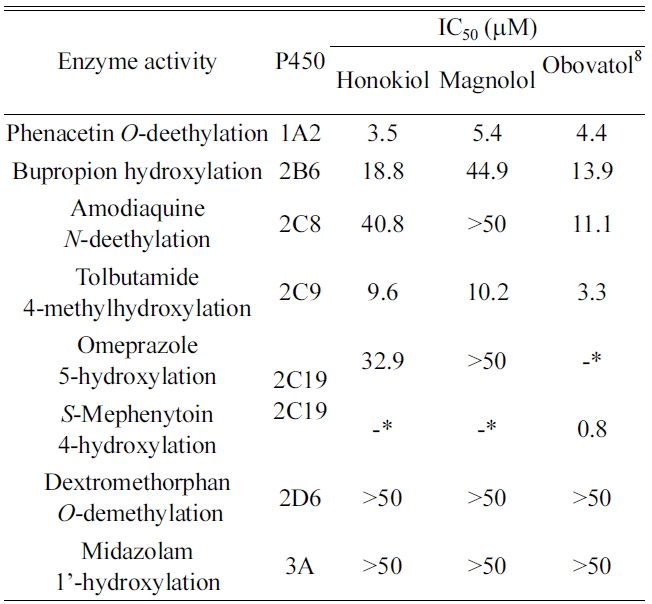
Inhibitory potency of honokiol and magnolol on seven P450 activities in human liver microsomes
0.2 μM. These
Honokiol and magnolol had negligible inhibitory effect (IC50 > 30 μM) on CYP2C19-mediated omeprazole hydroxylase activity whereas obovatol showed strong inhibitory potential (IC50 = 0.8 μM) against CYP2C19-mediated
Honokiol and magnolol at a concentration of 40 μM did not affect the activities of CYP2C8, CYP2D6, and CYP3A isoforms. These findings suggest that clinical interactions between these compounds and P450s such as CYP2C8, CYP2D6, and CYP3A would not occur.
In the present study, we investigated the inhibitory effect of honokiol and magnolol using human liver microsomes. Honokiol and magnolol moderately inhibited the metabolism of CYP1A2-mediated phenacetin