Recent developments in MS-based glycomics and glycoproteomics have rapidly advanced the field and pushed the boundaries of glyco-analysis into new territories. This review will lay out current workflows and strategies for characterization of the glycoproteome, including (in order of increasing complexity and information content) preliminary site mapping, compositional glycan profiling, isomer-specific glycan profiling, glycosite-specific glycopeptide profiling, and finally, glycoproteomic profiling.
Complex carbohydrate chains, known as glycans, decorate most of the human proteome and modulate crucial biological activities ranging from cellular signaling to protein folding.1?4 Glycosylation may be found on a variety of medically- and pharmaceutically-significant proteins, including biologic drugs (such as monoclonal antibodies or erythropoietin),5,6 vaccines (e.g. viral proteins hemagluttinin and neuraminidase),7,8 and clinical biomarkers (including cancer markers CA 15-3, CA-125, and PSA).9?11 Techniques for characterization of protein glycosylation, therefore, are in high demand by biopharmaceutical manufacturers, governmental regulatory agencies, and clinical laboratories alike.12,13
Unfortunately, glycoproteins are extraordinarily complex molecules. Unlike other bio-oligomers (such as nucleic acids or proteins), glycans are not synthesized by a template-driven process, but rather through interaction with a complex biochemical environment containing hundredsof glycosidases and glycosyltransferases.14 Understandably, glycoproteins often possess significant structural hetero- geneity, with glycoforms numbering in the hundreds or even thousands, and thus characterization remains a significant analytical challenge.12,13
To analyze complex mixtures (such as glycans or glycoproteins), multi-dimensional and/or high-resolution separation is necessary. Mass spectrometry (MS) neatly fits the requirements of glyco-analysis, as it is highly amenable to hyphenation with complementary technologies (such as tandem MS or LC) and, furthermore, is available in several high-resolution flavors (including TOF, FT-ICR, and Orbitrap).15 It should come as no surprise, therefore, that mass spectrometric methods are the cornerstone of modern glyco-analytical technology. This review will cover current MS-based workflows and strategies for comprehensive characterization of the glycoproteome.
Level 0: Glycosylation Site Mapping
As a precursor to glycoprotein characterization, scientists must first determine whether or not a protein is glycosylated. Commonly, a complex mixture of proteins or peptides (both glycosylated and unglycosylated) is screened for this possibility. From this mixture, glycoproteins or glycopeptides are enriched by either lectin affinity16?20 or hydrazide chemistry.21?24 Various methods of enzymatic or chemical deglycosylation may then be applied to remove the glycan,12,25 leaving a scar of some sort at the protein attachment site. For example, glycoprotein/-peptide digestion by peptide N-glycosidase F (PNGase F) replaces glycan-attached asparagines with an aspartic acid, causing a mass difference of +0.9858 Da. The deglycosylated proteins or peptides can be analyzed by MS as part of a standard proteomics workflow, with the mass difference caused by the scar indicating the presence of glycosylation at a particular site.26,27
More recently, several reports have suggested the use of isotopic labeling to increase the specificity (and therefore accuracy) of N-glycosite mapping techniques.28,29 In this modified procedure, glycoproteins or glycopeptides are digested by PNGase F in 18O water, such that the glycan-attached asparagine is deaminated into an 18O-labeled aspartic acid. The resulting mass difference of +2.9890 Da is readily distinguished from glycosite-obfuscating peptide modifications such as spontaneous deamidation (+0.9858 Da) and can thus be definitively associated with the presence of a glycosylation site.
Though glycosylation site mapping is relatively straight- forward, the information content of the experiment is limited, as all glycan information (compositional or structural) is lost once the glycan moiety is removed. Therefore, to acquire a more comprehensive picture of the glycoprotein(s) in question, researchers often complement glycosylation site mapping with various glycan profiling techniques.27
Level 1: Compositional Glycan Profiling
Glycans, whether N- or O-linked, can be released from proteins by a variety of enzymatic or chemical methods, as discussed in the previous section. Following release, the released glycans may be enriched and analyzed. One of the simplest ways to analyze released glycans is to profile them according to mass, and therefore, composition. Briefly, this involves ionization of a heterogeneous glycan mixture,
separation of glycan ions according to (exact) mass, and finally, quantitation of each detected glycan ion mass. Each mass would correspond with some composition of monosaccharides; thus, the term “
In the early days of glycomics, researchers were often faced with the issue of limited MS sensitivity. Glycans are naturally polar, and therefore harder to ionize due to low volatility. To compensate, researchers came up with derivatization strategies that increased glycan hydro- phobicity, either by the addition of a hydrophobic tag (e.g. 2-AB labeling)30?32 or by replacement of polar groups with nonpolar ones (e.g. permethylation).33?35 While these derivatization strategies are still in use today, they are no longer necessary due to vast sensitivity improvements in modern mass spectrometers. Accordingly, native, label-free glycans are now the analyte of choice for comprehensive glycomic profiling.35,36
As an analyte, native glycans are not only easier to prepare, but also enable greater analytical sensitivity. While glycan derivatization techniques are continually being optimized, they never quite reach 100% efficiency, and so side reactions or incomplete reactions always occur at low levels.37,38 The unwanted reaction byproducts obfuscate the resulting mass spectra and, consequently, raise the noise level. In contrast, when native glycans are analyzed, noise in the mass spectrum originates primarily from electronic interference inside the MS detector, rather than from any chemical byproducts. In the past decade, decreases in instrumental noise levels have far outstripped decreases in chemical noise levels, and thus native glycan profiling today provides far greater sensitivity than previously imagined.6,39,40
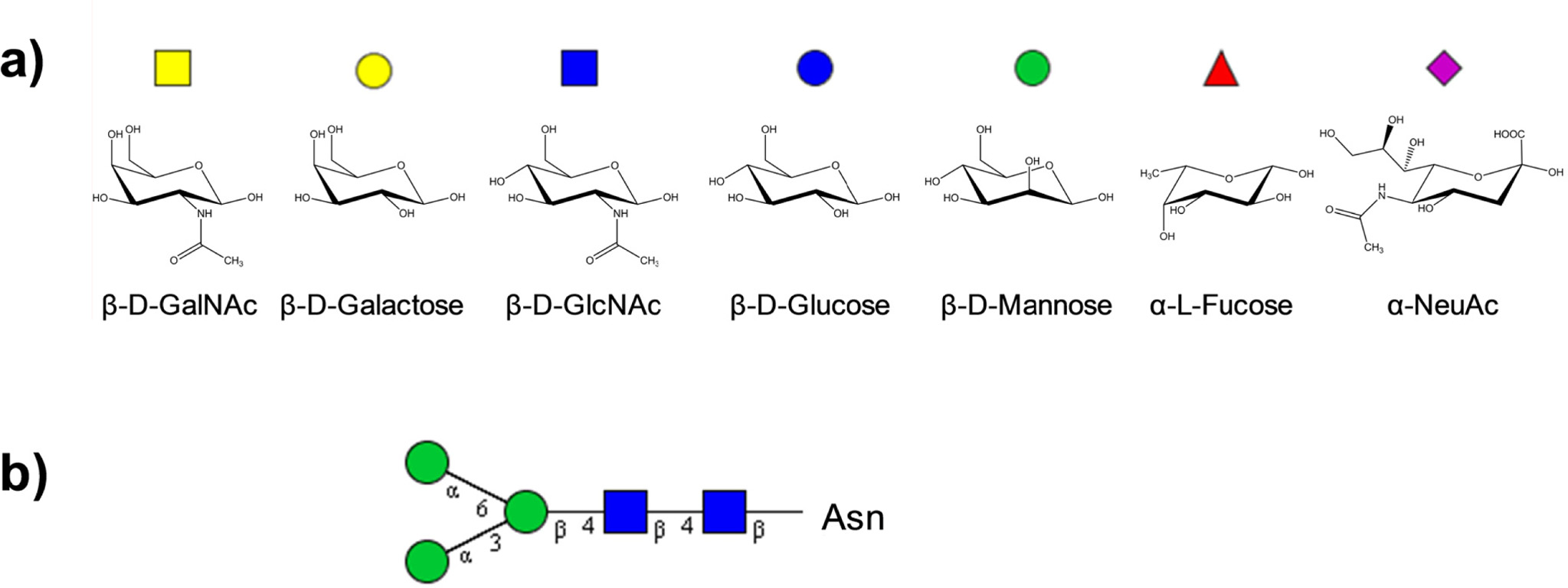
Mass spectrometry (specifically, high-resolution MS) is an ideal analytical technology for compositional glycan analysis. In comparison to other bio-oligomers, glycans are easily separated and differentiated by mass. Human glycans, for example, are built from just four monosaccharide masses: hexose (including glucose, galactose, and mannose), at 162.0528 Da; N-acetylhexosamine (including N-acetylglucosamine and N-acetylgalactosamine), at 203.0794 Da; fucose, at 146.0579 Da; and N-acetylneuraminic acid, at 291.0954 Da (Figure 1(a)). With sufficiently high
MS-based glycan identification can be further refined by incorporating biological filters into mass-matching algo- rithms. The core structure and biosynthesis pathways of N-glycans, in particular, are well-defined (Figure 1(b)), allowing implementation of sensitive and specific biological filters that rapidly screen mathematically-possible monosaccharide combinations and accurately eliminate biosynthetically-impossible glycans.41 O-glycans, unfortunately, are not as structurally well-defined, but are still subject to some biosynthetic rules that can aid glycan mass-matching.43,44
Biological filters, in conjunction with experimental evidence, can be used to create glycan mass libraries.45 These libraries comprise a list of all possible (or probable) glycans found within a certain glycome: for example, human serum; or recombinant erythropoietin; or even bovine milk. Experimental MS results can then be matched to the libraries, and peaks automatically identified. Such libraries have already been applied successfully to various cancer biomarker studies39,46 as well as biopharmaceutical batch-to-batch comparisons.6
Level 2: Isomer-specific Glycan Profiling
A large part of glycan structural heterogeneity stems from isomeric glycans. Whereas sequence is sufficient to determine the primary structures of linear bio-oligomers such as nucleic acids and proteins, glycans are branched, with many possible linkages and configurations amongst the component monosaccharides. Analytical technology has only recently achieved the ability to differentiate and profile isomeric glycan structures, and some studies have already been able to correlate differences in the isomeric configuration of glycans with concrete biological effects.8,47,48 However, many of the effects of glycan isomeric configuration remain unknown and ready for discovery by the next generation of young glycobiologists.
In order to identify glycan isomers, one must first understand their origin. Glycan isomers occur due to a) connectivity, when monosaccharides are connected in different orders; b) branching, when monosaccharides are attached to different antennae; and c) linkage, when monosaccharides are linked at different carbons on the sugar molecule. Depending on the cause of the isomerism, different analytical technologies can help elucidate precise glycan structures.
The first step towards glycan structural elucidation is isomer separation. While single-stage MS can easily separate glycans of different compositions, it obviously cannot separate those with identical compositions (and masses) but different structures. Luckily, MS can easily be hyphenated with other structure-sensitive technologies, such as ion mobility spectrometry MS (IMS-MS) and/or LC/MS.15
Structure-sensitive LC/MS is currently the most popular method of separating and differentiating glycan isomers. This method utilizes structure-sensitive stationary phases, commonly amide49,50 or porous graphitized carbon.39,40,46,51?55 In addition to the glycan composition, these stationary phases respond to the three-dimensional structure of a glycan. As a result, different glycan isomers are retained by the stationary phase for different lengths of time. Upon elution, the glycans are ionized by electrospray and detected in the mass spectrometer. The abundances of different glycan isomers can then be quantified and compared.46,56
Glycan structural libraries enable rapid glycan identification by structure-sensitive LC/MS. Using these libraries, glycan structures can be identified by a combination of exact mass and reproducible (or time-calibrated) retention time. Since glycans are separated and identified in not just one, but two dimensions (namely, mass and retention time), identification is highly specific. Glycan structure/retention time libraries are currently available for human serum glycans as well as human milk oligosaccharides, with many more glycome-specific structural libraries in development stages.52,53
For laboratories equipped with the requisite technology, LC/MS/MS spectral databases can provide even higher specificity in glycan differentiation and identification. Following structure-sensitive LC/MS, glycan isomers may be separately fragmented by tandem MS techniques such as collision-induced dissociation (CID) or electron transfer dissociation (ETD). Different glycan isomers fragment differently according to their three-dimensional structure,and thus display different fragmentation patterns. Each distinct fragmentation pattern serves as a spectral “fingerprint” for a particular glycan isomer.57 Some fragments can even serve as diagnostic ions for the presence of certain glycan structural motifs, such as bisecting N-acetylglucosamine (GlcNAc), polylactosamine, or N-glycolylneuraminic acid (NeuGc or NGNA).58,59
Other techniques are currently available for separation and/or differentiation of glycan isomers. These include MSn, in which glycans are sequentially fragmented until isomer-specific glycan fragments are found;60,61 IMS, in which glycans are separated according to cross-sectional area;62?64 and exoglycosidase digestion, in which glycans are sequentially digested by enzymes that act only upon specific glycosidic linkages.65 Each technique comprises a unique method of glycan structural differentiation and/or separation, and would be highly informative in complement to structure-sensitive LC/MS. Unfortunately, none of these techniques can currently be performed in a high-throughput or automated manner, making them of limited use for rapid screening in clinical or industrial settings. Nevertheless, their raw potential is high, and future developments are expected to transform these technologies into viable options for glycan isomer-specific profiling.
Level 3: Glycosite-specific Glycopeptide Profiling
While glycan profiling provides an abundance of information about changes to the glycosylation machinery, it does little to explain the effect of glycosylation on glycoprotein structure, function, or recognition. In order to discern the biological effect of altered glycosylation, one must localize these alterations to the originating glycoprotein and, more specifically, the originating glycosylation site.66?68
Glycoproteins are often decorated with multiple glycans, each attached at a different amino acid site on the protein. The glycans can be compositionally and structurally similar, or different, and can even be a combination of N- and O-glycans. In order to determine which glycans are attached at which sites, the site itself must be analyzed with the glycan still attached. Thus, site-specific glycan profiling necessarily uses glycopeptides as the principal analyte.
Glycopeptides are generated by digesting glycoproteins with proteases. This serves two purposes: 1) Increasing ionization efficiency and analytical sensitivity by decreasing the size of the analyte; and 2) Isolating glycans on separate molecules, in order to simplify site assignment. Ideally, following protease digestion, each glycopeptide should contain only a single glycan and a single site of glycosylation. Thus, identification of the peptide moiety and the glycan moiety would be sufficient for localization of the glycan to the glycosylation site. (In contrast, if a glycopeptide contained only a single glycan but multiple sites of glycosylation, an analyst would have significant difficulty determining which site of glycosylation the detected glycan was attached to.)
Trypsin is often used, particularly by those trained in classical proteomics, to digest glycoproteins into glycopeptides.19,69?72, Due to the high specificity of tryptic digestions, the resulting tryptic peptide masses can easily be predicted
Other proteases or protease mixtures have also been used for glycoproteolysis.73?76 One popular alternative to trypsin is pronase, one of a large class of nonspecific or “multi-specific” proteases.38,54,77?84 Pronase has multiple specificities and activities that enable it to cleave glycoproteins at a variety of sites, creating glycopeptides with (generally) shorter peptide moieties. Therefore, using pronase, it is much easier to isolate just one glycan and one glycosylation site onto each glycopeptide. Additionally, the smaller glycopeptides are more amenable to LC isomer-specific separation and subsequent MS detection.85,86
Glycopeptides can be prepared and analyzed by MS in much the same way as glycans, discussed earlier. However, MS data interpretation is much trickier for glycopeptides than for glycans, due to the high number of compositional “building blocks” that can be found in a peptide moiety- in humans, up to 19 different amino acid masses (in addition to the four monosaccharide masses found in the glycan moiety, discussed previously).
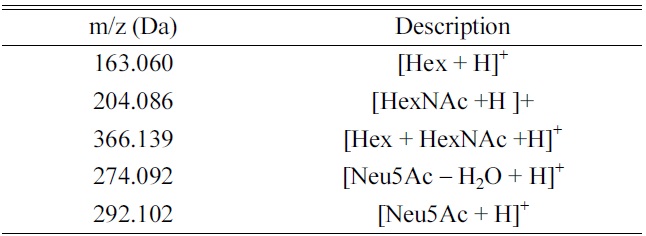
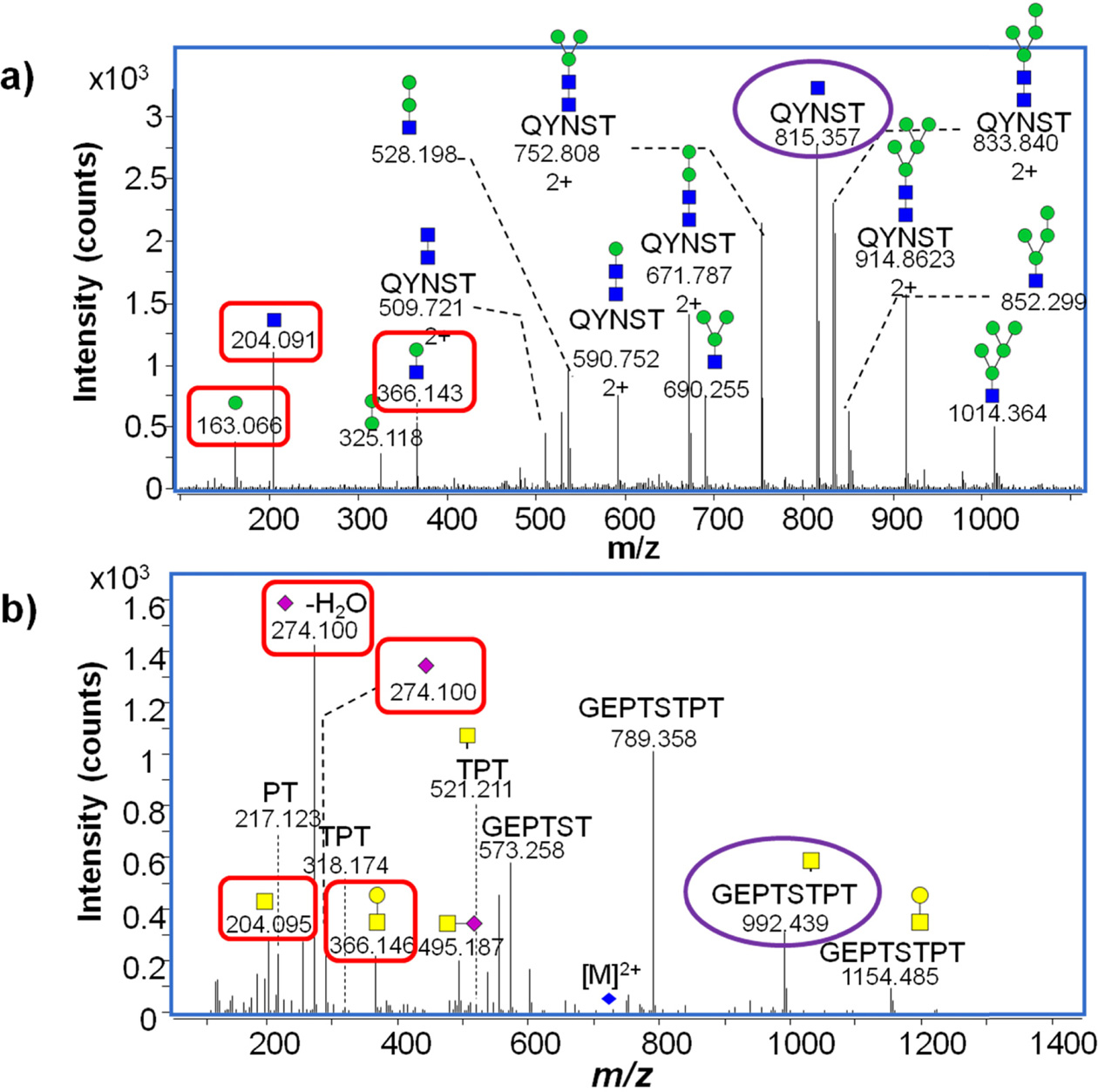
CID MS/MS fragmentation of glycopeptides produces a number of diagnostic fragment ions that can be used to not only confirm that the precursor is a glycopeptide, but also compositionally identify the glycopeptides (Table 1). In positive mode, common indicators of a glycopeptide precursor include
[Table 1.] Diagnostic fragment ions.
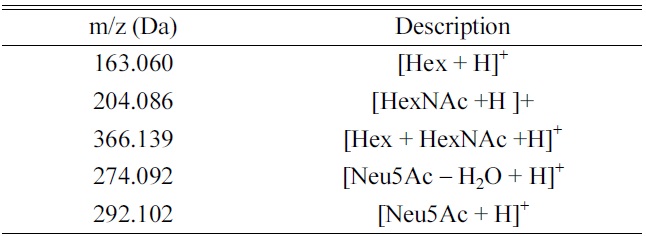
Diagnostic fragment ions.
of glycosidic bonds (vs peptide bonds) these fragments are almost always found in relatively high abundance in CID MS/MS spectra of glycopeptides. Other fragments are diagnostic of certain glycan moiety types- for example,
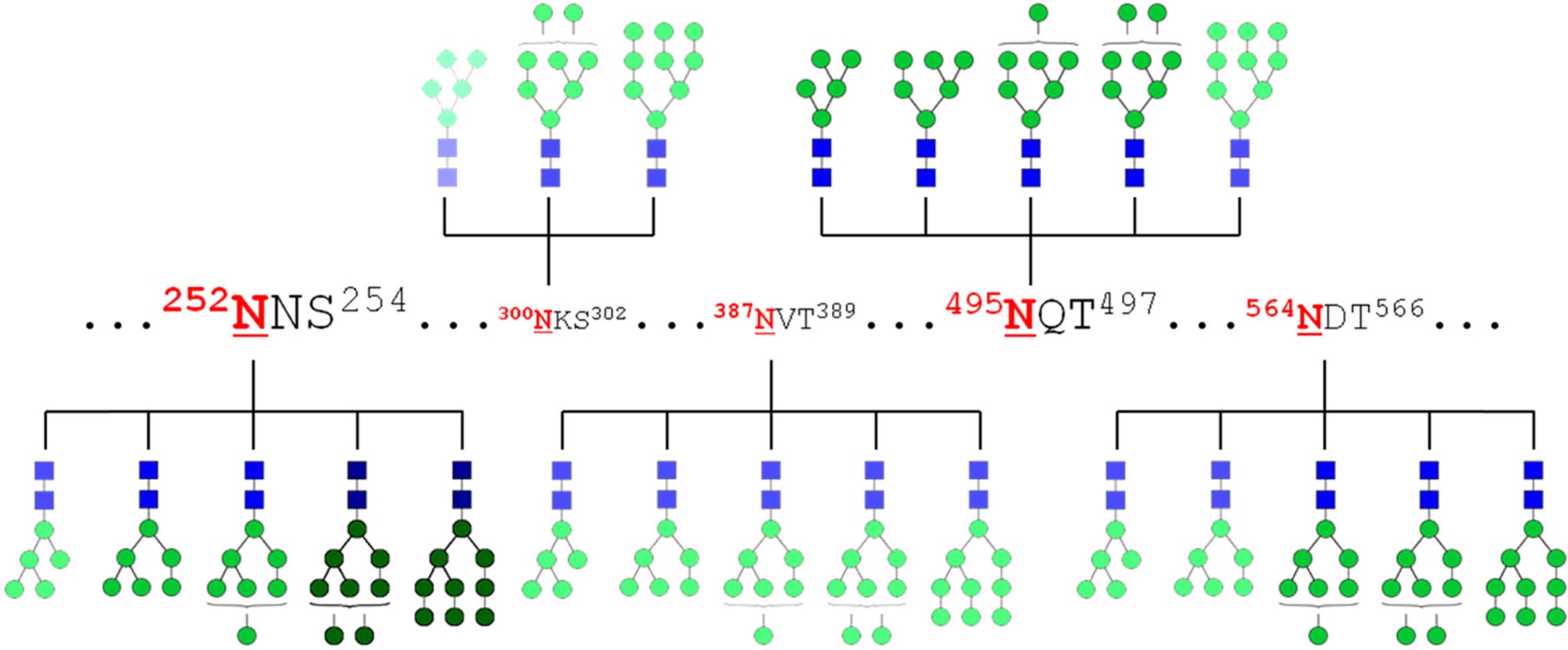
Glycosite-specific glycopeptide profiling enables the creation of glycosylation maps such as those shown in Figure 3. Depending on the equipment and methodology used, these maps can be quantitative, or merely qualitative. Application of these maps to biomarker studies enables researchers to identify specific protein glycoforms that vary with disease, providing targets for clinical screening or diagnostic tests.
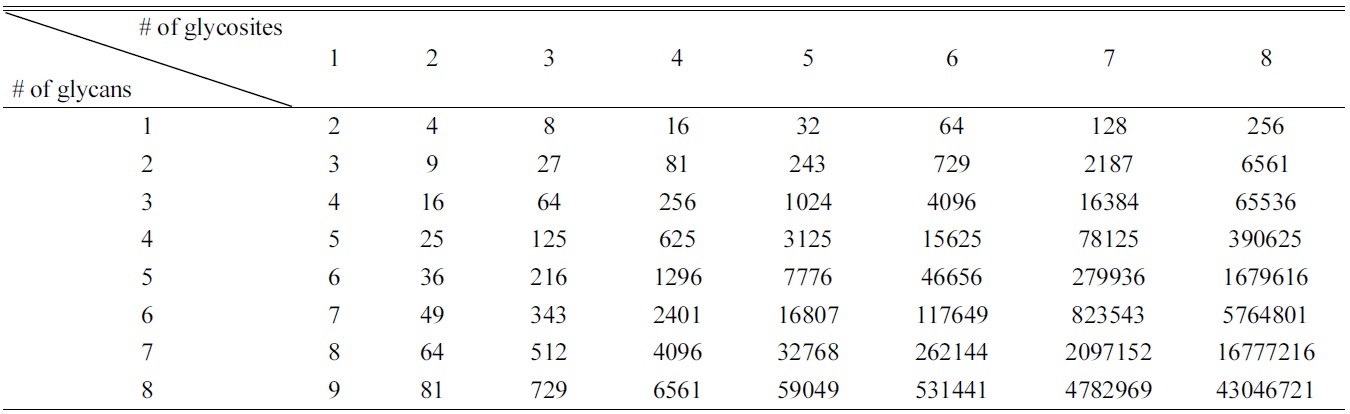
[Table 2.] Possible glycoforms according to number of glycosites and glycans.
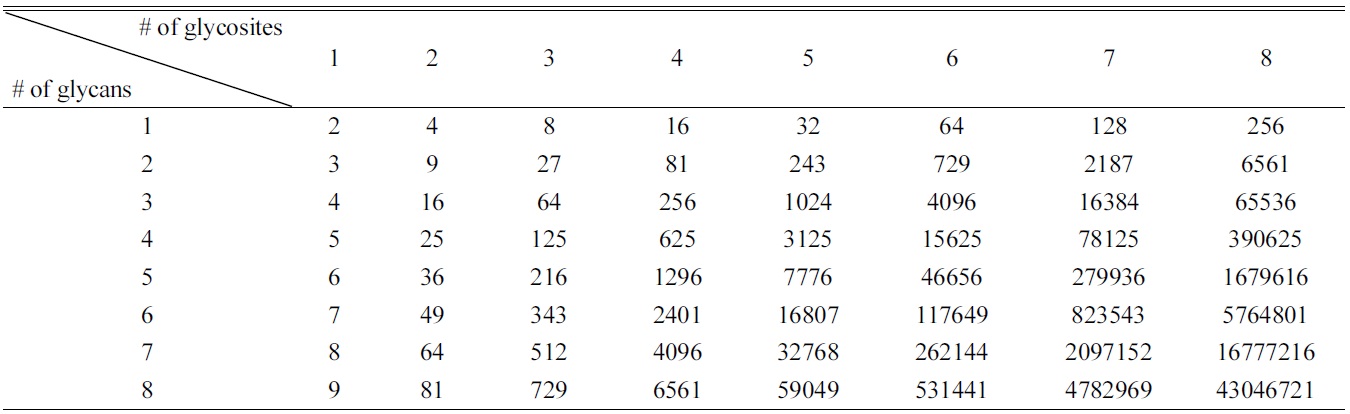
Possible glycoforms according to number of glycosites and glycans.
The term “glycoproteomics” is evocative of the highly-automated computer algorithms currently being used in proteomics workflows. However, despite all of the recent advances in glycopeptide profiling, discussed in the previous section, automated site-specific profiling of whole glycoproteomes is still beyond the reach of current technology.
Part of the issue is data content. Though we often speak of glycoproteins as a single entity, they are each in fact a heterogeneous population with identical peptide sequences but very different attached glycans. A theoretical glycoprotein with four sites of glycosylation and five possible glycans at each site could have (5 + 1)4 = 1296 different glycoforms (including the possibility of no glycosylation of each glycosylation site). Glycoforms multiply rapidly if additional glycosites or additional glycans are found to be possible (Table 2): in the aforementioned case, just one additional glycosite and one additional possible glycan would give (6 + 1)5 = 16807 different glycoforms.25 While computerized search algorithms can be programmed to match experimental and theoretical glycopeptide masses, the sheer number of theoretical masses means that almost any random contaminant or noise peak could potentially be matched to a theoretical glycopeptide. Without further data filtration, the false discovery rate skyrockets, severely decreasing the probability of accurate assignments.
To solve this problem and turn glycoproteomics into a real possibility, researchers will need to develop methods to finely fractionate whole glycomes. Immunoaffinity-based methods already exist that extract specific glycoproteins from complex mixtures so that they can be profiled in detail.92 However, for broad-scale application, these methods will need to be supplemented (or supplanted) with more general methods of separation: chromatography and electrophoresis. Multidimensional separation has already been applied to proteomics with great success,93?95 and is just now starting to be applied to glycoproteomics.96 Further developments in this field could enable automated, site-specific profiling of whole glycoproteomes in the near future.
Developments over the next few years in mass spectrometric strategies and bioinformatics will lead to comprehensive analysis of glycoproteins. Structure-specific glycomics is now within reach and just starting to be applied to biomarker research as well as biotherapeutic characterization. Meanwhile, advances in glycoproteomic analysis are beginning to yield quantitative strategies for the characterization of site-specific glycosylation. These new strategies will help us gain a greater understanding of an important yet largely unexplored portion of our biology: the glycome.