Expressions of particular genes in microbial strain or community during utilization of different carbon sources involve a complex genetic network and differ with type and complexity of carbon sources. Such network can be studied by applying transcriptional profiling (gene expression), proteomics profiling (protein expressions) and metabolomics(metabolite mapping). The importance of fungi in providingthe foundation for our understanding of cell cycle, secretion mechanism, mitochondrial evolution, cellular pathways, and many more critical biological processes made us to sequence several fungal genomes; while issues like global warming, environmental pollution, green house gases emission, depleting fossil fuel has made us to focus on renewable bioresources, such as the use of fungal secretomefor lignocellulosic biorefinery. Again, the non-food feedstocksuch as agricultural residues (e.g., corn stover, sugarcane bagasse, husks, rice and wheat straws, barley straws etc.), agricultural processing byproducts (e.g., corn fiber, sugarcane bagasse, seed cake, etc.), energy crops (e.g., switch grass, poplar, banagrass, miscanthus, etc.), hardwood, softwood, cellulosic waste, forest waste and municipal solid wastes are abundant renewable biomasses that can serve as a major substrate for lignocellulosic biorefinery and bioenergy. Thus, genomes of well known lignocellulose degrading fungi belonging to Ascomycota (
Fungi digest or degrade complex substrates outside the cell before their intake and hence they secrete large number of extracellular hydrolytic enzymes, generally arbitrated as secretome. The secretome includes all secreted proteins, either anchored to cell surface or in the extracellular environment and also the proteins involved in secretory pathway. These secretory enzymes have been investigated from different point of views or due to their biotechnologicalapplications.9 Traditionally, colorimetric methods were usedfor quantitative analysis of extracellular secreted enzymes. However, these methods when evaluated based on their limitations such as range, sensitivity, specificity, ease of conducting assay, ability to detect isoforms, reagent cross-reactivity, and inability to quantify each constituent protein of complex secretome emphasized development of alternative methods.
Extensive advances in technology, emergence of genomics, proteomics and protein-tagging technology made it possibleto determine the complex composition of the secretome. Proteomics provides a global view of the protein expressed during biomass degradation process and, in combination with other omics technologies, has potential to uncover biomass degradation mechanism, and thereby advance the development in designing of enzyme cocktail for efficient biomass hydrolysis. To degrade biomass, fungi secrete vast number of extracellular enzymes hence secretome related studies could be more relevant in understanding underlying biomass degradation. Therefore, this review paper provides an up-to-date review on recent developments on proteomic techniques used to study lignocellulolytic enzymes and biochemical route that fungi use to degrade plant biomass; and attempts to shed light on the fundamental understandingin lignocellulosic biomass hydrolysis.
Overview of Lignocellulolytic Fungi
Fungal biotechnology has already established societal benefits by bringing innovative and valuable healthcare and food products to market. Despite progress in green chemistry and proven contribution of fungi in recycling lignocellulosic biomass, production and commercialization of commodity products such as chemicals, fuels or electricity from lignocellulosic wastes is still lagging. The recycling of photosynthetically fixed carbon by microbe, particularly, by fungi is fundamental biological process and integral part of biogeochemical cycle. Fungi degrade biomass via different mechanism and differ in the way they make cellulose, hemicelluloses accessible to enzymes. Based on the morphology arising from the attack (white or brown pockets) they are broadly divided into white rot, brown rot and soft rot. Two distinguishable patterns of biomass delignification such as simultaneous delignification and selective delignifi- cation have been documented.7 White rot fungi are numerous and include members of both ascomycota and basidiomycota. These white rot fungi have potential to completely degrade lignocellulosic biomass since they are capable to degrade lignin by cleaving bonds between Cα and Cβ. While, brown rot prevalently attack on coniferous wood, removes cellulose and hemicellulose and can not degrade lignin or degrade only small part. Thus, both white and brown rot are very specific trait of basidiomycetes. Fungi like
Lignin removal opens the way for wood colonization by other microbial populations which is a key step for the recycling of carbon. Lignin degradation is an oxidative process where fungal extracellular peroxidases and oxidases(glyoxal oxidase, pyranose-2 oxidase, and aryl-alcohol oxidase) oxidizes the polymer. Enzymes such as lignin peroxidase (LiP), manganese peroxidase (MnP) and versatileperoxidase (VP) by
Gel-Base Separation and Proteomic Analysis of Secretome
Protein separation techniques including gel electrophoresis or chromatographic separation prior to mass spectrometry analysis play major role in proteomics. Polyacrylamide gel electrophoresis (PAGE) separation technique is used as a preparatory technique for separation or purification of proteins prior to identification by MALDI-TOF or LC-MS/MS analysis, while chromatographic methods have been coupled with MS using both on-line and off-line separation steps. Gel electrophoresis differentiates molecular entities depending on their physical characteristics such as size, shape, charge and isoelectric point. Two-dimensional PAGE(2D-PAGE), a classical proteomic technique that separates proteins by isoelectric point and molecular weight, remain as a well established technique in proteomic analysis of complex samples. The general work flow involves protein separation by 2D gel, protein visualization by staining, gel image analysis, protein spot excision, tryptic digestion, analysis of the peptide mass, database search and further validation.21
Application of 2D in Fungal Proteomics
Due to potential to promote economical and environmentalfriendly sustainable energy production, plant feedstocks are at the forefront of the biofuel industry. Several structural and compositional features hinder hydrolysis of lignocellulosic cellulose to sugars and other organic compounds and hence several physical, chemical and enzymatic pretreatments have been tested. The advantages of enzymatic pretreatment and its environmental friendly nature attracted more research on fungal lignocellulolytic enzymes. Using most popular 2D electrophoresis (2DE) technique and mass spectrometry as the core tool, extracellular proteins of
Using 2DE, Hori and coworkers28 investigated the effects of xylan and starch on protein secretion by the basidiomycete
LC-Based Separation and Proteomic Analysis of Secretome
For comprehensive identification of proteins, mostly, a peptide-centric approach is adopted where proteins are trypsin digested, and then components of the peptide mixture are separated by using liquid chromatography prior to its MS analysis. Such LC-based proteomics techniques are much more important to produce unambiguous protein identification since samples contain thousands of proteins and its tryptic digestion results hundreds of thousands of peptides. Electrostatic repulsion hydrophilic interaction chromatography (ERLIC) has been developed and further compared with SCX.34?36 Further, Adav et al.37?40 applied this ERLIC technique to study secretome of the lignocellulolytic fungi with a particular objective to explore expression of lignocellulolytic enzymes for biorefinery.
The accurate protein quantitation is currently one of the most challenging and rapidly changing areas of proteomics. The choice of methods for quantitative proteomics depends on multiple factors including the source of the samples, the number of samples, the number of treatments being compared, the type of equipment available, time requirementand most importantly expenses. Several quantitation methodsincluding isotope labeling approaches like Isotope-Coded Afffinity Tag (ICAT), Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC), 15N/14N metabolic labeling, 18O/16O enzymatic labeling, Isotope Coded Protein Labeling(ICPL), Tandem Mass Tags (TMT), Isobaric Tags for Relativeand Absolute Quantication (iTRAQ), and other chemical labeling have been adopted in quantitative proteomics.41,42 The majority of the mass spectrometry quantitative methodsinvolves tryptic digestion of proteins and then takes advantage of measurements made on peptide level to compute a summarized value for corresponding protein. Such approachcan be divided into two categories, one is label free quantitation and other involves use of stable isotopes or isobaric tags. Again, quantitative proteomics can be subcategorized into absolute which estimates changes in protein expression in terms of an exact amount/concentration(i.e., nmoles/mmoles) of each protein present in the given sample; and relative quantitation that determines fold change(up- or down-regulation) of a protein relative to control.
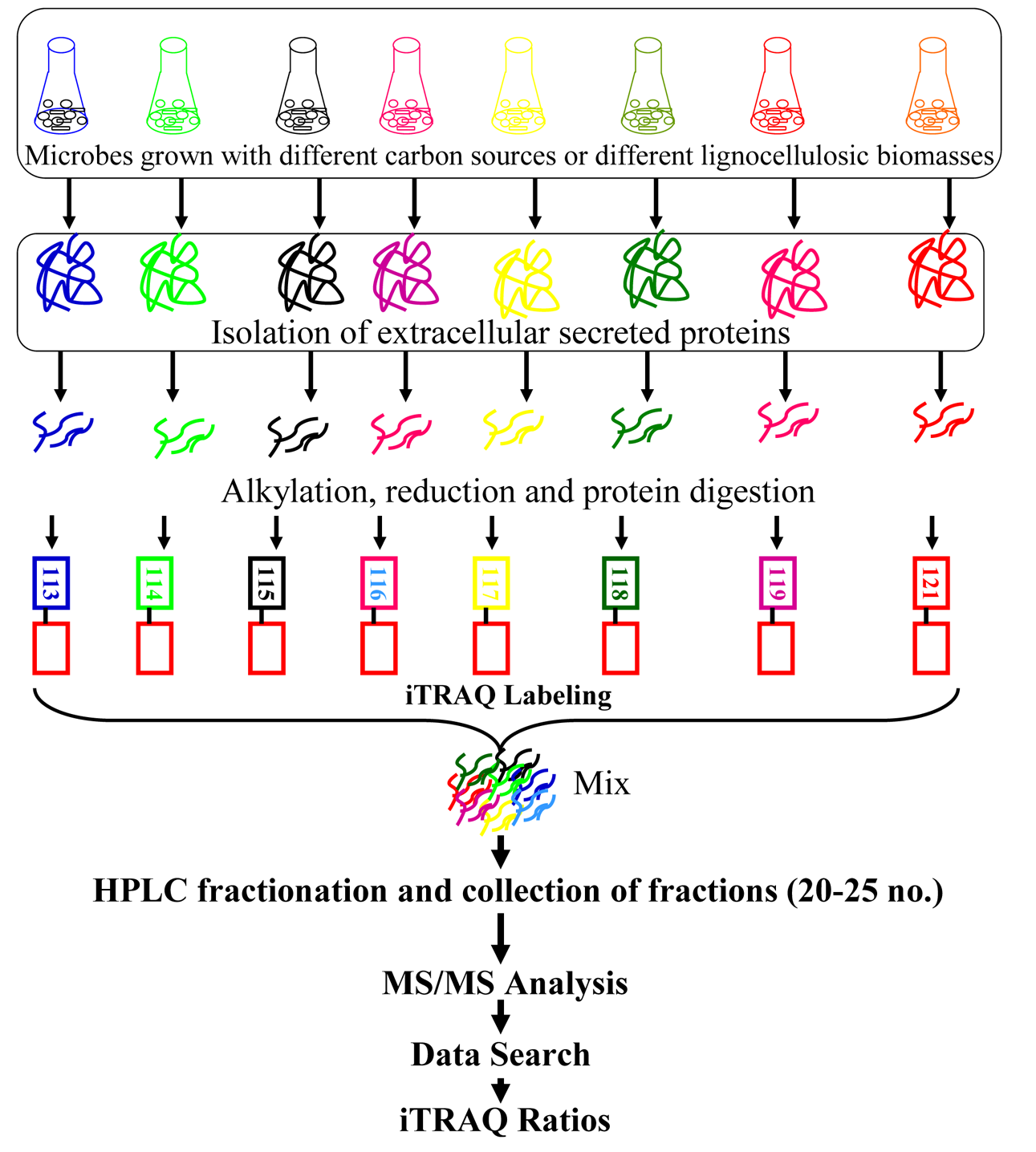
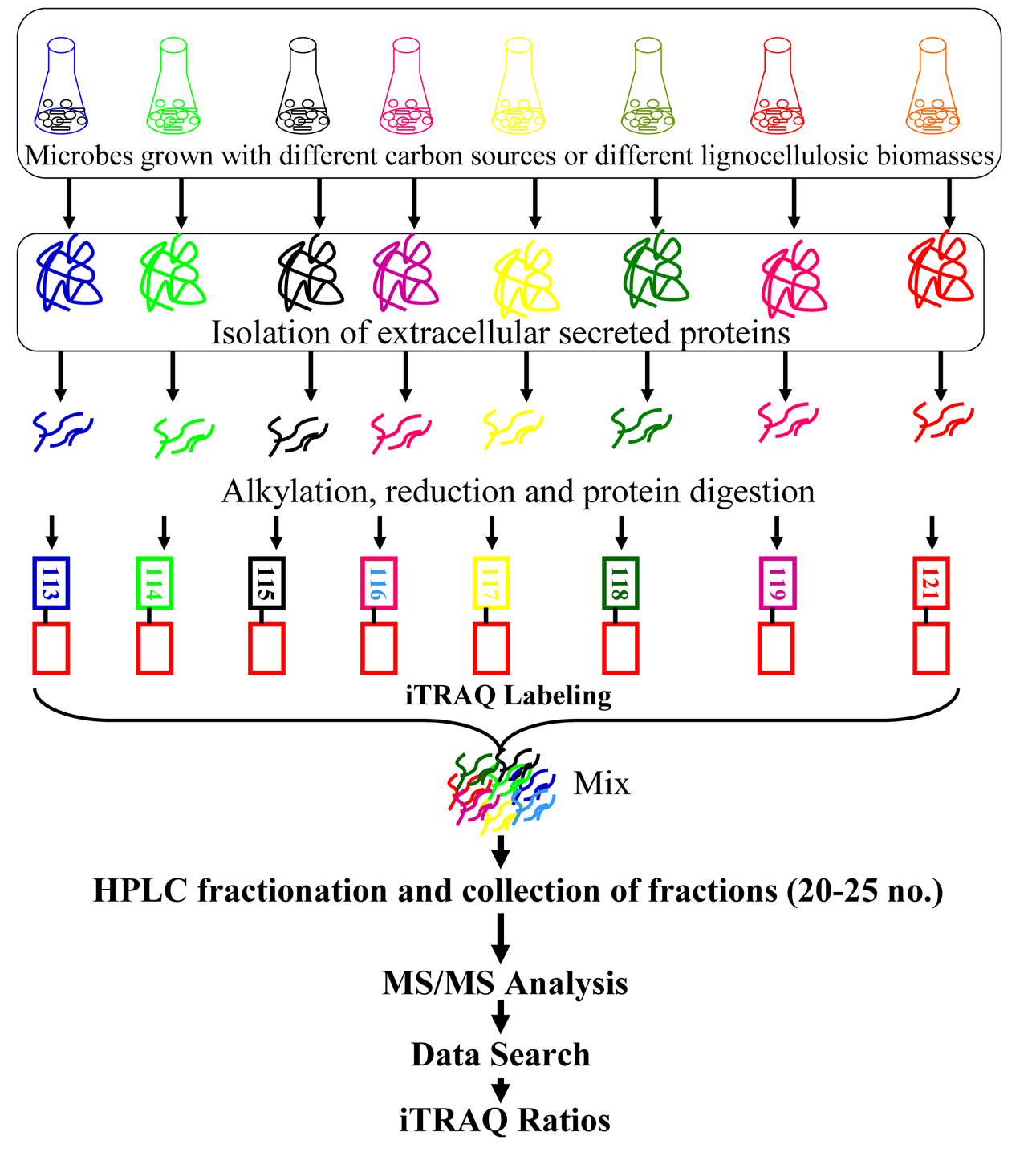
The iTRAQ technology utilizes isobaric reagents to label the primary amines of peptides and proteins and widely used in discovery-based proteomics. Due to multiplexing reagent designing, iTRAQ allows multiplexing of four to eight different samples in a single MS experiment.43,44 DuringiTRAQ-labeling isobaric tags react with amine groups in peptides generated by tryptic digestion and label N-terminal amino group and ε-amino group of lysine residues. Similarly, two different amine reactive versions of tags called tandem mass tags (TMT) are also commercially available.45 Since iTRAQ quantitation is at MS/MS level rather than MS level, it gives better results due to lack of chemical noise that interfere quantitation quality at MS1 level. Again, the tags used in iTRAQ reagent are isobaric, the signal in the mass spectrometer is the sum of the peptide contribution from all samples, so there is a gain in sensitivity.43
The iTRAQ technique has been applied to profile the quantitative expression of secretory lignocellulolytic enzymesof
The culturing of
wild
Manavalan et al.48 applied iTRAQ technique to profile expression of lignocellulolytic enzymes of basidiomycete
Although, iTRAQ technologies have many attractive attributes and widely used in proteomics, but yet this technique have several limitations. Wu et al.50 compared cICAT, iTRAQ, and DIGE, and found that iTRAQ was more sensitive for quantitation, but more susceptible to errors in precursor ion isolation, indicating mass spectrometerinterference hinders iTRAQ reliability. The issues like iTRAQ-reproducibility and reliability has been addressed by Chee et al.51 Minor disadvantages include incomplete labeling since the labeling reaction is pH dependent. This limitation can be eliminated by checking pH of the sample solutions before labeling. The major limitations of iTRAQ technologies are its precision, accuracy and reliability.52,43 The comparisons of the label free MSE quantification with the iTRAQ quantification using a QTOF instrument recorded divergence in the degree of upregulation of up to a factor of 9.2 between the two quantification methods, with the iTRAQ method giving lower ratios.53 Similarly, a discrepancy of underestimation of up to 5 factors was noted when investigating 8-plex iTRAQ reagent with analysis on a QStar XL instrument.54 The issues like underestimation of ratios, limited dynamic range (fold changes of < 2 orders of magnitude are typically reported), high reagent cost and a limitation for ion-trap instruments due to the “1/3rd rule” are critical and their possible solutions are discussed in several reviews.55?61 The ambiguity of measurements can be explainedat least in part by correlation with reporter ion signal intensity. However, several methods like intensity weighted averaging of multiple spectra of given peptide or protein, post-acquisition filtering of data to exclude low intensity measurements, data normalization and advances in ion transmission hardware may address the issues with accuracyand precision.52,62?64 A more persistent issue with isobaric tagging is that of accuracy. The most common reason for the accuracy issue is the presence of co-eluting peptides with m/z in the precursor ion isolation window.
Stable isotope labeling by amino acids in cell culture (SILAC), introduced by Mann and co-workers for high-throughput quantitative proteomic analysis has been used inmany inmany different organisms for protein levels quantification.65
SILAC has been used to study the temperature-dependent expression of whole proteome of
Label-free quantification of fungal secretome
Although, stable isotope labeling methods are powerful for accurately determining changes in protein level but suffer from limited dynamic range. Quantitative proteomics with iTRAQ or stable isotope coding requires expensive labeling reagents, large amount of sample proteins and complex labeling procedures that reduces detection sensitivity. On the contrary, label free quantitation is free of labeling reagent; applicable to all type of biological samples, can process any number of samples, and can be a rapid protein quantitation techniques. Label free quantitation methods uses Exponentially Modified Protein Abundance Index (emPAI) that offers relative quantitation of the proteins in a mixture based on protein coverage by the peptide matches in a database search result.72 The spectral counting method that relies on counting the number of spectra that map to a given protein across multiple LC-MS analyses, peptide peak intensity, the peak area, the height, quantification of the MS signal and average TIC are the other documented approaches under label free quantitation.73 By taking advantage of the linear correlations between peak areas and corresponding peptide abundance,
iTRAQ or label free quantitation?
The comparison of iTRAQ and label-free quantitative approaches found that iTRAQ gave 54 % single peptide identifications, while only 0.9 % of the identifications in label-free methods were single peptide hits.53 Mostly data with single peptide remain as a low confidence and if all single peptide hits preferred to be excluded during quantifications then the impact on proteome coverage by iTRAQ would be considerable. This study indicated that label free quantitative techniques are better than iTRAQ-quantitative approach. Again, in a study carried by Phillip Wright group on a
Combine quantitative and label free approach
In lignocellulosic biomass hydrolysis as well as in other biological systems, determination of the exact concentrationof proteins and time resolved changes are important to uncover the possible mechanism. Hence, by combining iTRAQ-quantitative and label free quantitative approach, protein abundances in the secretome of individual fungal strains and constructed microbial communities during lignocellulosic biomass hydrolysis have been reported.39 By combining quantitative data from iTRAQ and label free emPAI approach, authors determined the abundances of lignocellulolytic proteins in the secretome of
Lignocellulosic biomass comprising cellulose, hemicelluloseand lignin is virtually inexhaustible most abundant renewablecarbon resource. The enzymatic conversion of cellulosic, forest and agricultural wastes to fermentable sugars can potentially generate significant amounts of bioenergy to mitigate global warming and current global energy crisis. In addition to bioenergy, lignocellulosic biomass deconstructionwill produce value added products and chemicals. Applicationof proteomics in profiling quantitative expressions of extracellular secreted fungal proteins during biomass deconstruction provides a glimpse into lignocellulolytic enzyme action and may divulge promising shift from bio-prospecting and novel enzyme discover. Proteomics study of fungal or microbial community secretome highlighting quantitative protein abundances in the secretome would shed light on the designing of microbial enzyme cocktail for biomass hydrolysis. Thus, recent efforts to explore lignocellulolytic protein expression during biomass degradation by different proteomic techniques have been presented. We also propose the following perspectives to the potential development of the proteomic techniques for fungal enzymes analysis and novel fungal enzyme characterization in the near future.
An enthusiasm about the potential application of proteomics to discover enzymatic mechanism of biomass hydrolysis has been tampered by complexity, wide range of variably regulated and dynamic nature of enzymes. We discussed above several proteomics methods including quantitative and label free but yet little work has been done on absolute quantitation of each protein in the fungal secretome. To design efficient biomass degrading enzyme cocktail, absolute quantities of enzymes are very much important, and hence more research work on absolute
quantitation is required. Majority of published literature highlights only cellulases, hemicellulases and lignin degradingenzymes ignoring other enzymes such as chitinases, proteases, peptidases, transporters etc. Considering complex nature of biomass, quantitative proteomic profiling of whole secretomeenzymes, their discussion and interpretation, enzyme synergisms must expand beyond cellulases, hemicellulases and lignin degrading enzymes. Work on isolation of thermoresistant lignocellulolytic fungi, proteomic profiling of such themostable enzymes and novel candidate is warranted.