It is well known that the properties of polyacrylonitrile (PAN)-based carbon fiber are strongly determined by the nature of the precursor [1-4]. Considering structural homogeneity and char yield, homopolymers are highly sought raw materials [2]. Owing to outstanding molecular cohesive energy (237 cal/cm3), however, the fiber exhibits poor flexibility. Moreover, upon preoxidation, PAN precursors obtained from homopolymers tend to release heat at a rapid speed, thereby increasing the likelihood of the fiber being broken. In general, a second or third monomer such as acrylic ester, itaconic acid, or acrylamide (AM) is added to acrylonitrile (AN) monomer in order to change the cyclization reaction mechanism from a free radical cyclization reaction to an ionic reaction. The exothermic reaction of preoxidation can then proceed at lower temperature and at a low rate and an increasing number of construction units are transformed into char in a graphite-like structure, given that carbon fiber is an inorganic polymer fiber, and more than 90% is carbon. The content of char in the obtained fiber should be enhanced as much as possible in the form of a graphite-like structure after high-heat treatment. The enhanced formation of char can endow the obtained precursor fiber with higher homogeneity and compactness, and provides the the resultant carbon fiber with better mechanical properties [3]. Therefore, enhancement of char-yield in the polymer is necessary to obtain high performance carbon fiber.
However, in copolymerization, owing to the different reactivity ratio between two monomers, the copolymer components vary with the development of copolymerization [4,5]. In particular, in heterogeneous precipitation polymerization, the difference in solubility between acrylonitrile and comonomer in an aqueous phase and oil phase, as well as the difference in adsorbability to polymer particles, make the copolymer more polydisperse. The nonhomogeneity of a copolymer prepared by copolymerization also reduces the efficiency of initiation of acylamino. Furthermore, an excessive amount acylamino is detrimental to the final properties of carbon fiber. Therefore, if an appropriate amount of prepared acylamino is uniformly distributed on the surface of the copolymer chain, it will effectively initiate the cyclization reaction. Note also that there have been few studies on the enhancement of char by modification of homopolyacrylonitrile instead of copolymerization.
In addition to copolymerization, there are other ways to introduce functional groups into a macromolecule chain, such as acidic or alkaline hydrolyzing ?CN into ?CONH2 or COOH, etc. [6]. Such hydrolysis is carried out in a polymer solution under stirring. Since the conditions are uniform, the generated functional groups are well-distributed in the macromolecule chain. Alkaline hydrolysis is too intense, however, to control and acidic hydrolysis at high temperature also suffers from the same problem. According to Loevy et al. [7], if PAN hydrolysis is carried out in 65% HNO3 at 15-50℃, binary copolymers containing only AN and AAm groups are formed and the initial reaction rate is slow. However, the neighboring AAm groups have an accelerating effect [8], which is not desirable for the preparation of a copolymer to be used to prepare carbon fiber, as a random copolymer is desired. Therefore, PAN hydrolysis in 65% HNO3 at low temperature in a short time period is recommended.
The purpose of this study is to enhance char yield in AN-AM copolymer by acidic hydrolysis of homopolyacylonitrile. To this end, 1) homopolyacylonitrile was prepared by precipitation polymerization in a H2O/dimethyl sulfoxide (DMSO) solvent; 2) nitrile groups on the homopolymer were acid hydrolized at 23℃ in short time to introduce acylamino groups; and 3) characterization of the hydrolized copolymer in relation to increased char yield in the AN-AM copolymer was carried out.
AN (Beijing Chemical Reagents Co.) was freed from inhibitors by distillation at 76-78℃ before polymerization. α,α’- azobisisobutyronitrile (AIBN) was purified by recrystallization. DMSO and deionized water (H2O) were used as polymerization solvents. Sixty five percent HNO3 was used as a polymer solvent and a hydrolysis reagent.
2.2. Precipitation polymerization
Precipitation homopolymerization of AN was carried out in a four-necked glass reactor by adding AIBN as a catalyst, H2O/DMSO (40/60 wt/wt), and AN (25 wt%) on the basis of the reactions medium) for 2 h at 60℃ under a pure nitrogen atmosphere. After polymerization, a sufficient amount of distilled water was added into the reactor to precipitate the produced polymer. The precipitated polymer was filtered by a buchner funnel, washed with distilled water and ethanol successively, and dried under a vacuum at 40℃ for 48 h, and a homopolyacrylonitrile powder was then obtained.
Acid hydrolysis of nitrile groups in the homopolyacrylonitrile was performed at 23℃ in 65% nitric acid. A set of 2 g homopolyacrylonitrile powder was dissolved in 98 mL 65% nitric acid and maintained at 4℃ for 47 h with slow stirring, where hydrolysis can practically be ignored [9]. At suitable intervals, the hydrolyzed polymers were precipitated and washed with cold water and dried to constant mass [10]. In order to distinguish different samples effectively, the homopolyacrylonitrile obtained from homopolymerization is denoted as P0, and the modified AN-AM copolymers are labeled P1, P2, P3, and P4 corresponding to 0, 5, 13, and 18 h, respectively. Specifically, P1 was obtained immediately at the end of dissolution and no longer maintained at 23℃, but there was still a slight change of its structure, even though the degree of hydrolysis was relatively low.
2.4.1. Infrared spectroscopy
Composition change in the copolymers prepared by acidic hydrolysis of homopolyacrylonitrile was verified by Infrared spectroscopy (D/max2500VB2+/PC Fourier transform infrared spectrometer) using a resolution of 4 cm-1 and 32 scans per sample.
2.4.2. X-ray diffraction
X-ray diffraction (XRD) patterns of the copolymers were obtained with an X-ray diffractometer (XRD-7500, Rigaku, Japan) at 40 KV and 40 mA with Ni-filtered CuKa radiation (λ = 1.54 A).The scanning speed was 4°/min and the scanning step was 0.02°. Crystallinity was calculated by Hinrichen’s method.
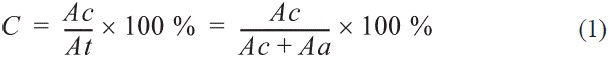
where At is the total peak area, Aa is the amorphous peak area, and Ac is the crystalline peak area.
2.4.3. Differential scanning calorimetry
Differential scanning calorimetry (DSC) curves were obtained from a differential scanning calorimeter (NETASCH, STA449c) from 25 to 400℃ at a heating rate of 10℃ /min-1 in nitrogen.
2.4.4. Thermal gravimetric analysis
A thermal gravimetric analysis (TGA) was carried out by using a NETASCH device, model TG 209F3, by heating samples from 50 to 900℃ in nitrogen at a heating rate of 10℃ /min-1, or changing the gas between air and nitrogen and heating rate.
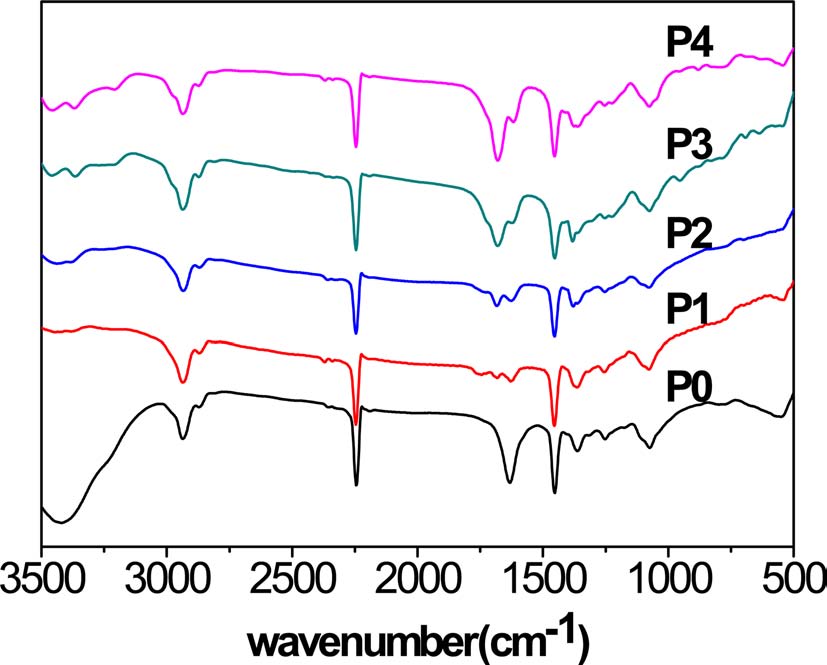
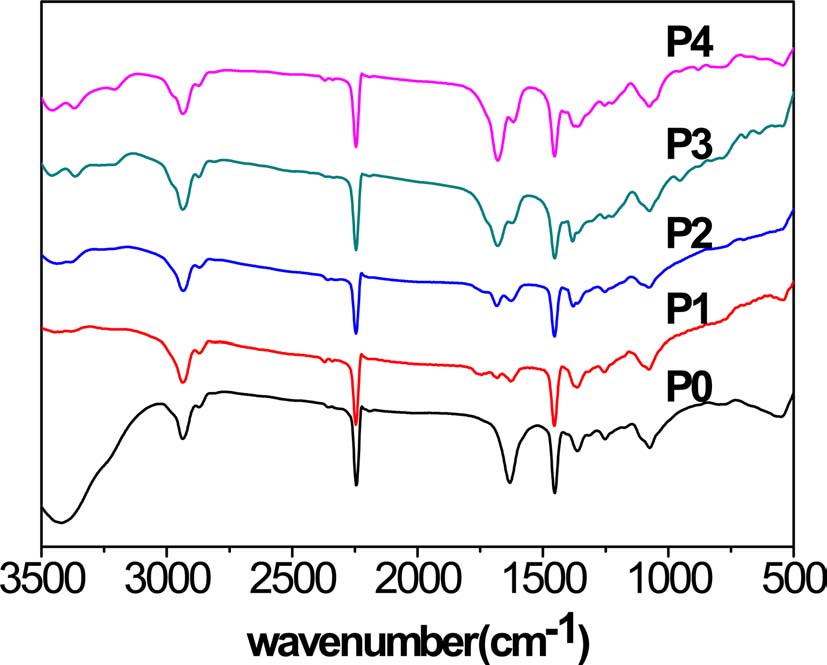
The infrared spectra of the homopolyacrylonitrile and ANAM copolymers showed higher absorption bands at the wave numbers 1452 cm-1 and 2240 cm-1, corresponding to ?CH2 and ?C≡N vibrations, respectively, which can be used to show the decrease in the nitrile group content through a comparison of
the two bands, although the decline was not very obvious. New bands were observed at 1621 cm-1, 1678 cm-1, 3368 cm-1, and 3454 cm-1. The bands at 1621 cm-1 and 1678 cm-1 correspond to ?C=O and ?NH2 vibrations of ?CONH2, respectively. Those at 3368 cm-1 and 3454 cm-1 correspond to symmetric and asymmetrical telescopic vibration bonds of ?NH2, and generally appear together. The four bands became increasingly distinct and strong. However, there were no bands at the wave numbers 1734 cm-1 and 1173 cm-1 corresponding to ?C=O and ?C-O vibrations of -COOH, respectively (Fig. 1).
Based on the above results, it is suggested that there was an increasing proportion of acylamino due to acidic hydrolysis of the nitrile group, and there was no sign of a carboxylic acid group in the copolymers.
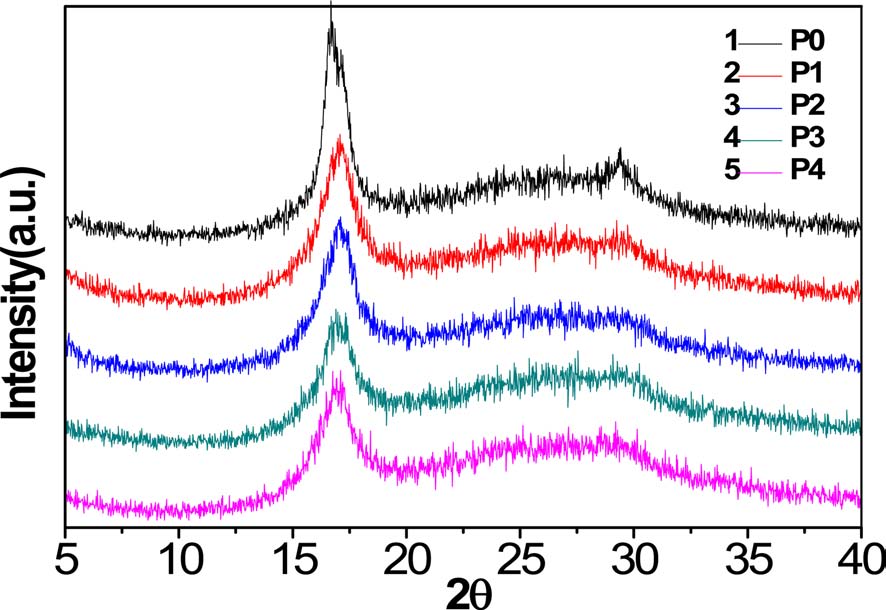
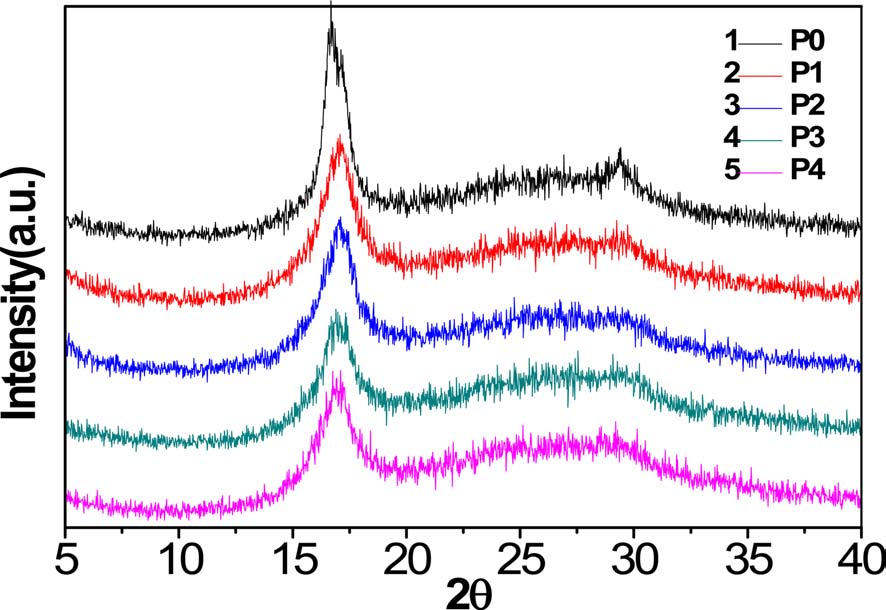
As can be seen from Fig. 2, XRD spectra of homopolyacrylonitrile and AN-AM copolymers showed sharp and strong diffractions at 2θ = 16.9° and weak diffractions at 2θ = 29.45°, corresponding to the (100) and (110) crystal faces, respectively [11]. A broad scattering profile near 24.5° indicates that there is an irregular molecular structure throughout the macromolecule [9].
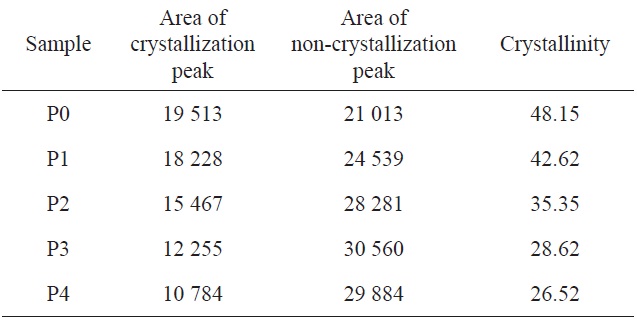
With increasing acidic hydrolysis time, the diffraction intensity of the (100) crystal face decreased and the (110) crystal face diffraction became increasingly less distinct. As shown in Table 1, the degree of crystallization of homopolyacrylonitrile was 48.15%, while the crystallinity of the copolymer decreased gradually and P4 decreased to 26.52%. It can thus be concluded that the crystallinity was inversely proportional to the content of acylamino.
There are intermolecular and intramolecular nitrile interactions due to CN dipole-pair bonds, and hydrogen bonds between the nitrogen of the nitrile group and tert-hydrogen, where the former is stronger than the latter [10]. As a steric hindrance, the introduced acylamino can separate the nitrile group in the macromolecular chain, and then reduce the interactions. As a result, molecular regularity and crystallinity are decreased, but flexibility of the molecular chain and drawability of the fiber made from these were enhanced, which is beneficial to increase spinnability.
[Table 1.] X-ray diffraction of P0 homopolyacrylonitrile and P1-P4 copolymers
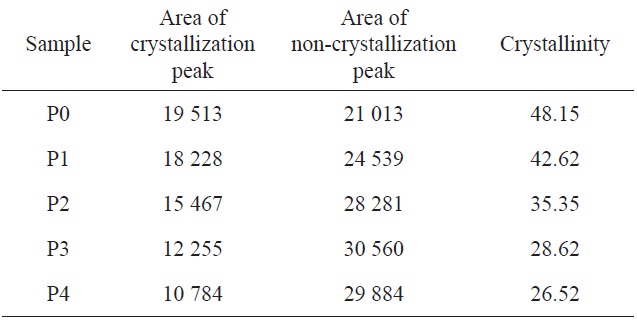
X-ray diffraction of P0 homopolyacrylonitrile and P1-P4 copolymers
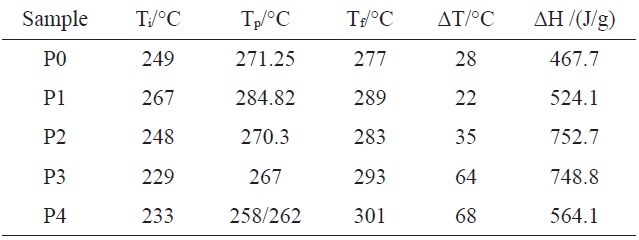
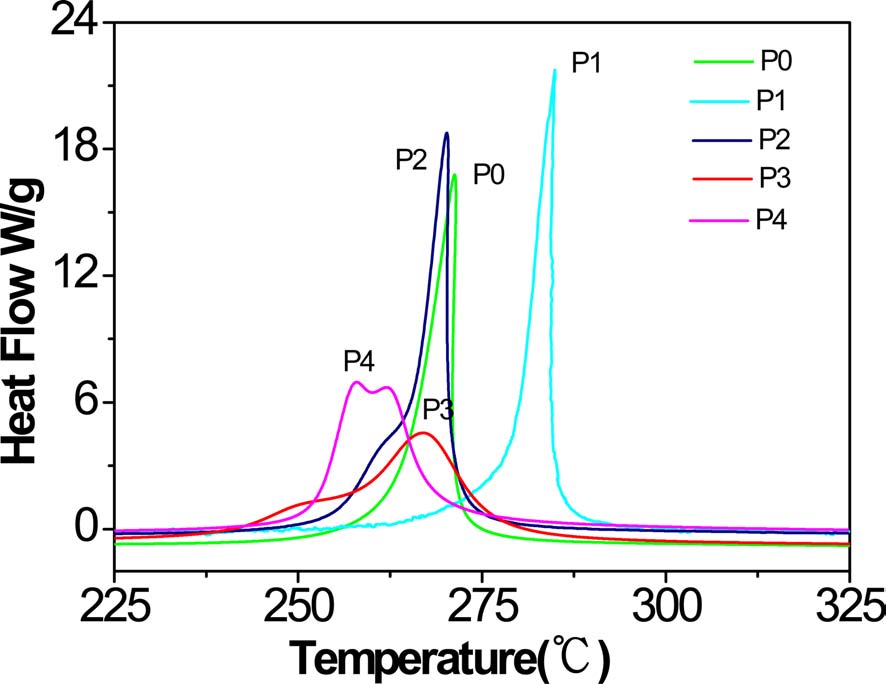
[Table 2.] Relative data of DSC from Fig. 3
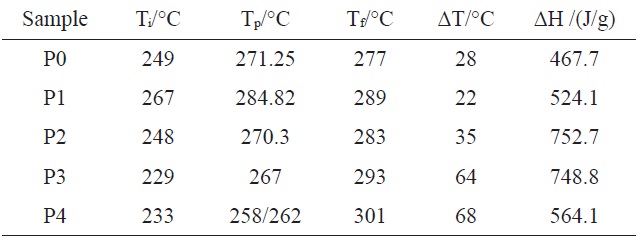
Relative data of DSC from Fig. 3
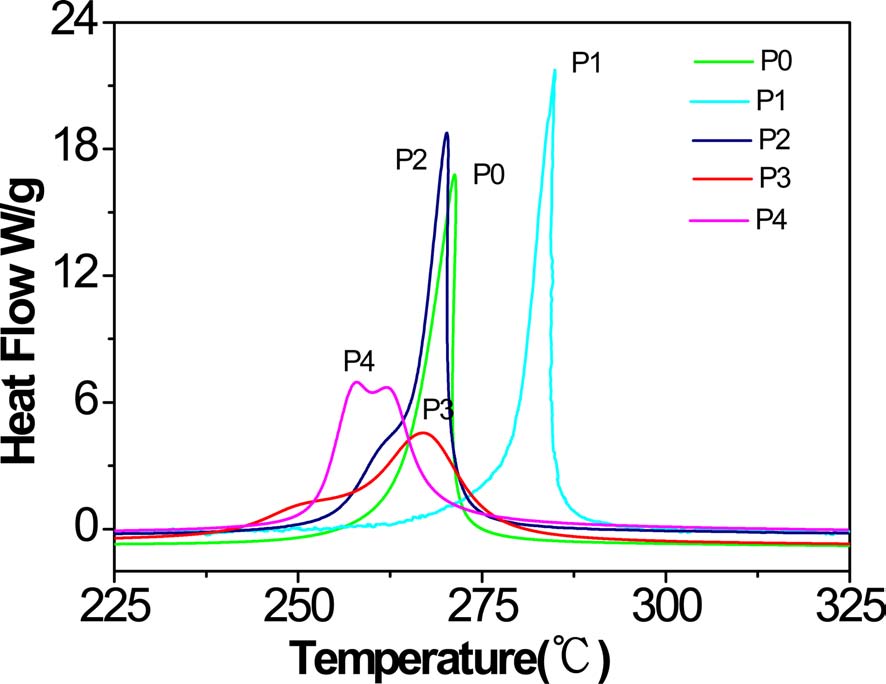
As can be seen from Table 2 and Fig. 3, the exothermic regimes of AM-containing PAN copolymers, except P1, were much broader and the cyclization reaction started at lower temperature, especially for P3 and P4, at 229 and 233℃, respectively, compared with P0 at 249℃. It was obvious thatermic peaks of P3 and P4 were lower and broader than that of P0, indicating thatermic reaction proceeded at a lower rate. Even more remarkable was that there were a couple of peaks for P3 and P4, and for P4, the first peak was higher than the second. It could be deduced that the introduced AM group partly changed the cyclization mechanism from free radical induced [12] to ion induced and slowed down the rate ofermic reaction. Zhang [13] and Kakida and Tashiro [14] also reported similar results.
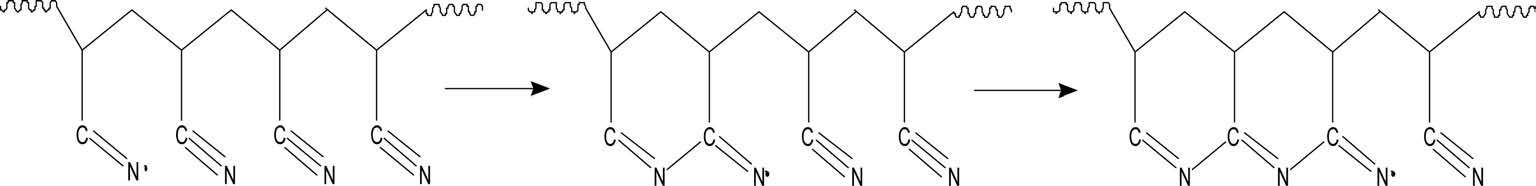
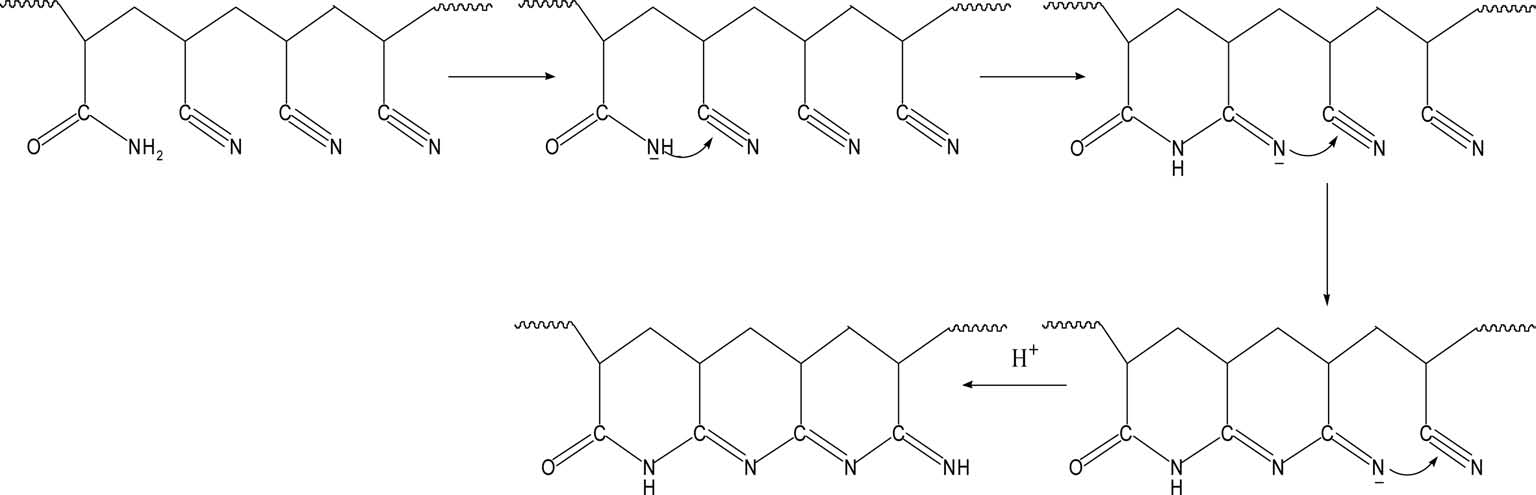
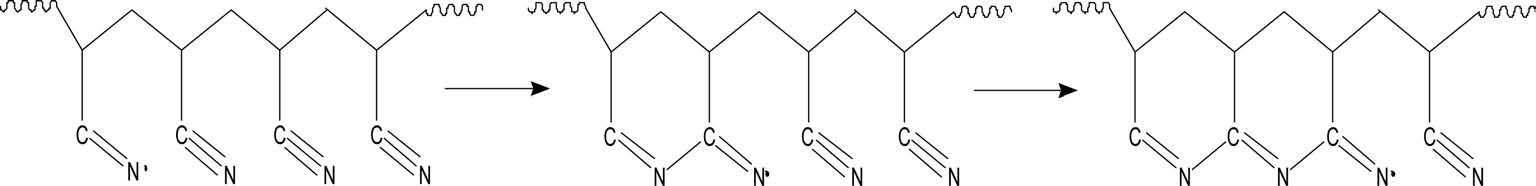
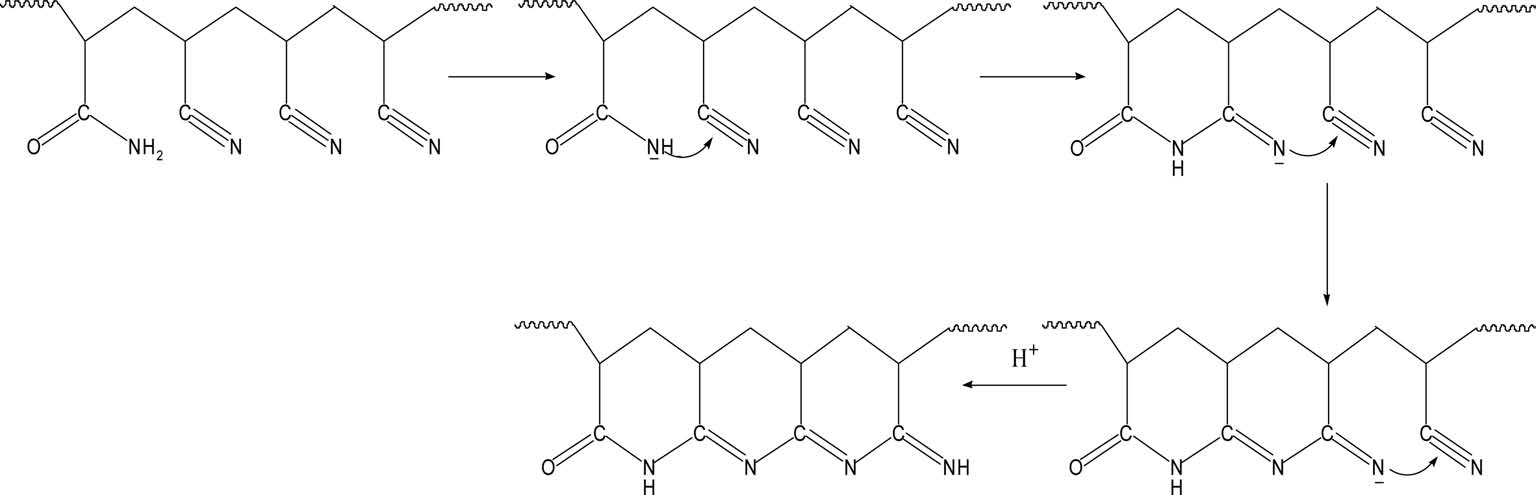
With regard to preoxidation, there are two mechanisms: a free radical cyclization reaction mechanism and an ionic cyclization reaction mechanism. It is generally acknowledged that preoxidation of homopolyacrylonitrile follows the free radical cyclization reaction mechanism (Fig. 4) and acylamino can activate the cyclization reaction towards the ionic mechanism (Fig. 5). With more acylamino, a higher proportion of preoxidation follows the ionic cyclization reaction mechanism.
More energy is needed to initiate the radical cyclization reaction than the ionic reaction. Once begins, it proceeds at a rapid rate. For the radical cyclization reaction, it begins at relatively high temperature and releases heat rapidly. As it is easier for acylamino to carry out a heterolytic reaction, the cyclization reaction can begin at low temperature and the reaction rate is slower than that of radical cyclization, which is beneficial for the formation of heat-resistant ladder structures and reaction control.
Sivy et al. [14] studied the change of AN/AM copolymer in the process of preoxidation in air by observation of its infrared spectrum, and suggested that the AM group can promote the cyclization and crosslinking reaction, leading to the formation of heat-resistant ladder structures. Kakida and Tashiro [13] studied the preoxidation of homopolyacrylonitrile fiber and copolyacrylonitrile fiber with a small content of AM or methacrylic acid, and suggested that comonomer acrylamide and methyl acrylic acid can accelerate both cyclization and dehydrogenation reactions. In this study, a similar conclusion could also be drawn through the DSC and TGA (Fig. 6) analyses, which showed that the AM group introduced by acidic hydrolysis can also accelerate cyclization and dehydrogenation reactions, producing more ladder structures with possible higher thermostability.
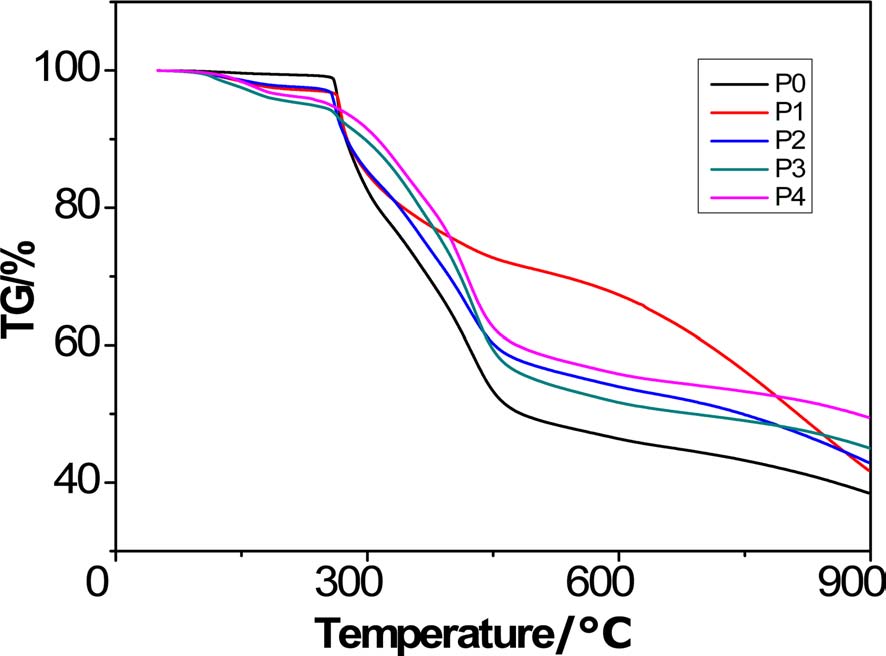
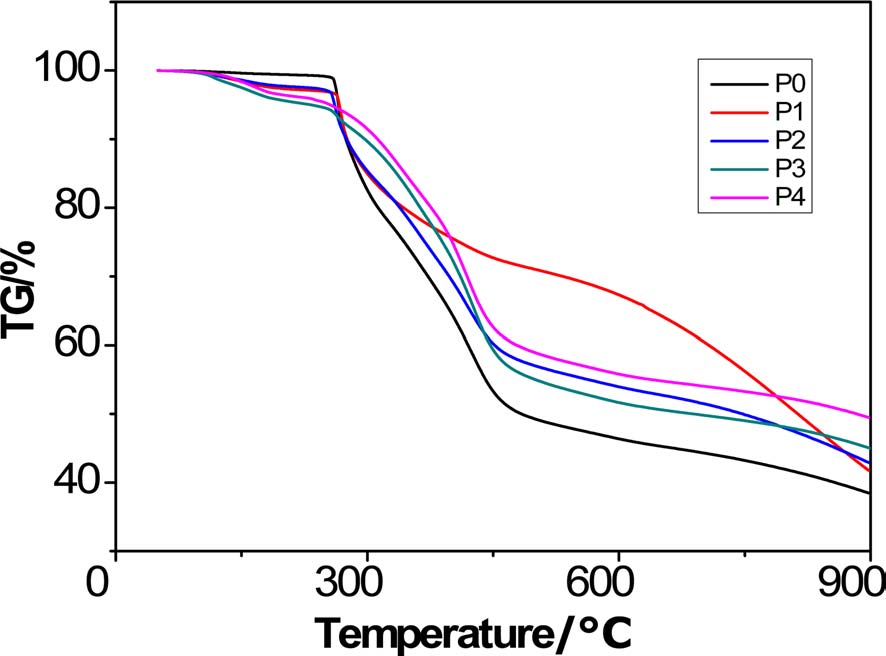
When the polymer is treated up to 900℃, carbon is the dominant component existing in the residue (usually over 90%). The resultant product after high-heat treatment is thus called char, and the mass fraction after heat treatment is accordingly called char yield. Therefore, the remaining mass fraction obtained at the end of the TGA analysis is referred to as char yield (Table 3).
To facilitate the analysis, the TGA curves can be divided into four regions: a trace weightlessness region, a region with rapid weight loss, a region with weight loss at a low speed, and finally a weightlessness platform region. There was a remarkable difference between the TGA curves of PAN homopolyacrylonitrile and the copolymers. In the TGA curve of homopolyacrylonitrile, slight weight loss can be observed before 265.5℃, but after 265.5℃ there was weightlessness at a very fast speed; this indicates fast reactions following the free radical cyclization reaction mechanism, and at this region heat was intensively released. In contrast, at low temperature, there was apparent weightlessness at a low speed for the copolymers, indicating that the introduced AM can initiate cyclization and dehydrogenation reactions at low temperature. Due to the transfer of mechanism of cyclization from a radical mechanism to an ionic mechanism, not only did the reactions proceed at a low speed, but the region with rapid weight loss became less obvious than homopolyacrylonitrile, indicating that the mass loss rate decreased greatly. In the region with weight loss at a low speed, the dominant reaction was breakage of
[Table 3.] Char yields from PAN homopolyacrylonitrile and copolymers

Char yields from PAN homopolyacrylonitrile and copolymers
chain segments. Owing to the AM group, more breakage of chain segments occurred at this region where the weightlessness of copolymers was greater than that of homopolyacrylonitrile. The height of the platform could reflect how much char was formed and the planeness of the platform could indicate the thermostability of the final product. With regard to the weightlessness platform region, it was noted that except for P1, the platform of copolymers was higher and flatter than that of homopolyacrylonitrile, thus indicating that the increased formation of ladder structures leads to higher thermostability in the PAN copolymers.
Overall, copolyacrylonitrile with higher char yield can be obtained by acidic hydrolysis of homopolyacrylonitrile.
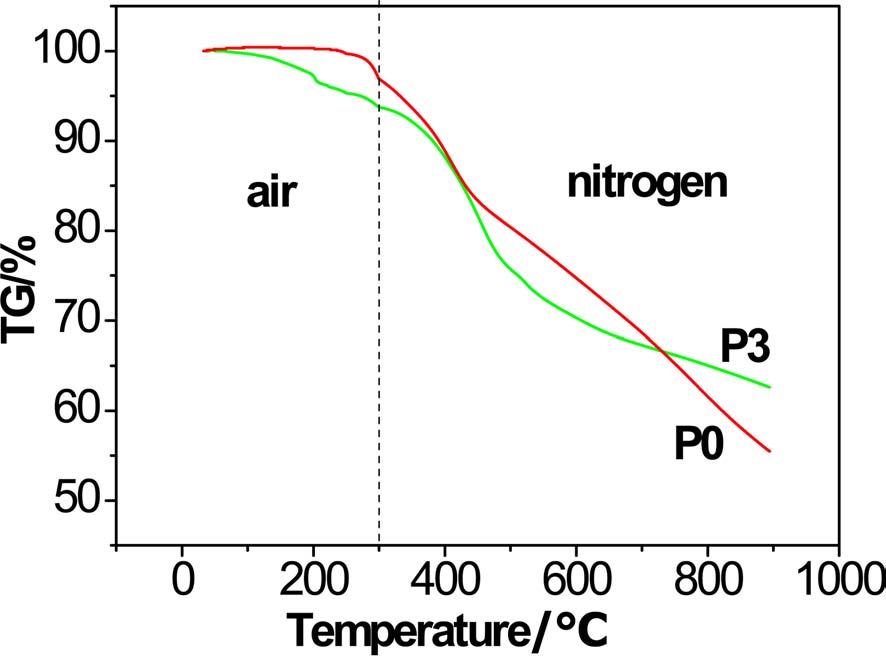
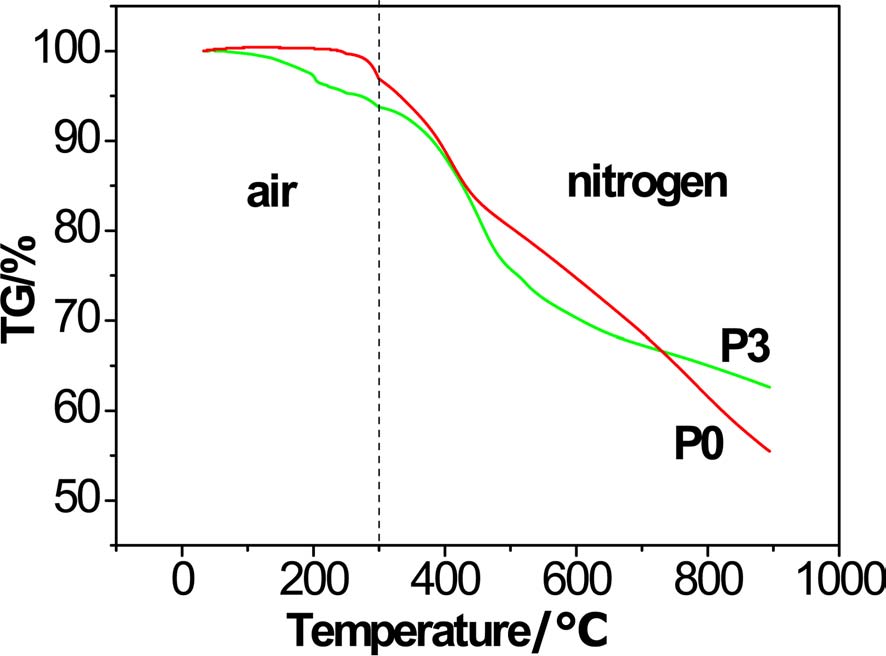
To verify the effect of increasing char yield, a special TGA test was designed to simulate the process of producing carbon fiber by changing the measurement atmosphere and heating rate. The heating rate was 20℃/min from room temperature to 200℃, 3℃/min from 200℃ to 300℃, and rate remained at 30℃/min over 300℃. At 300℃, the atmosphere was changed from air to nitrogen. From Fig. 7, with careful observation, it is seen that there was a slight gain of weight in the PAN homopolyacrylonitrile before 230℃, due to the oxidation reaction with air, and the maximum weight gain of PAN homopolyacrylonitrile was 0.4%. Weight loss of PAN copolymer occurred at the very beginning of the process. This is due the cyclization and dehydrogenation reactions being initiated at low temperature due to the already exited oxygen of acylamino. Although the thermostability of PAN homopolyacrylonitrile was higher than that of the PAN copolymer at low temperature, at higher temperature, the opposite trend occurred. At higher temperature, the homopolyacrylonitrile continued to lose weight at a rapid speed, but there was weightlessness platform for P3. The char yields were 55.43% and 62.60% for homopolyacrylonitrile and copolyacrylonitrile P3, respectively.
It might be concluded that, even in a practical process of preparing carbon fiber, there would be more carbon remaining from the raw material in the form of a graphite-like structure.
With the development of acidic hydrolysis, there was an increasing proportion of amide group, introduced by acidic hydrolysis of the nitrile group, leading to decreased crystallinity. DSC and a TGA indicated that the acylamino introduced by acidic hydrolysis initiated cyclization and dehydrogenation reactions at lower temperature, facilitating the formation of more ladder structures with higher thermostability. The maximum char-yield was 49.48% from the AN-AM copolymer at the experimental conditions, which was 30% higher than that of non-acidic hydrolysis of homopolyacrylonitrile (38.46%). Results from simulation of the practical process also showed an increase in the char yield and thermostability, where the char yields were 55.43% and 62.60% for homopolyacrylonitrile and copolyacrylonitrile P3, respectively, which further verified the enhancement of char yield. To sum, more carbon could be obtained from the raw material to form carbon fiber, which is beneficial to enhance the final product performance.