Chlorophenols (CPs) are widely used industrial chemicals that have been identified as being toxic to both humans and the environ-ment. Zero valent iron (ZVI) and iron based bimetallic systems have the potential to efficiently dechlorinate CPs. This paper reviews the research conducted in this area over the past decade, with emphasis on the processes and mechanisms for the removal of CPs, as well as the characterization and role of the iron oxides formed on the ZVI surface. The removal of dissolved CPs in iron-water systems occurs via dechlorination, sorption and co-precipitation. Although ZVI has been commonly used for the dechlorination of CPs, its long term reactivity is limited due to surface passivation over time. However, iron based bimetallic systems are an effective alternative for overcoming this limitation. Bimetallic systems prepared by physically mixing ZVI and the catalyst or through reductive deposition of a catalyst onto ZVI have been shown to display superior performance over unmodified ZVI. Nonetheless, the efficiency and rate of hydro-dechlorination of CPs by bimetals depend on the type of metal combinations used, properties of the metals and characteristics of the target CP. The presence and formation of various iron oxides can affect the reactivities of ZVI and bimetals. Oxides, such as green rust and magnetite, facilitate the dechlorination of CPs by ZVI and bimetals, while oxide films, such as hematite, maghemite, lepidocrocite and goethite, passivate the iron surface and hinder the dechlorination reaction. Key environmental parameters, such as solution pH, presence of dissolved oxygen and dissolved co-contaminants, exert significant impacts on the rate and extent of CP dechlorination by ZVI and bimetals.
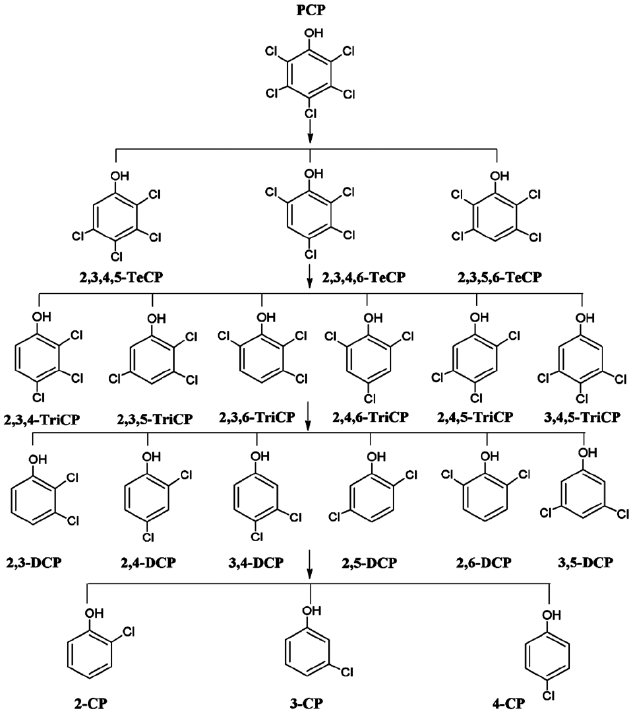
Chlorinated phenols (CPs) are industrial chemicals with wide applications, such as pesticides, herbicides, disinfectants, biocides and wood preservatives [1, 2]. The group of CPs com-prise of 19 different chlorinated phenolic compounds, includ-ing pentachlorophenol (PCP), 3 tetrachlorophenols (TeCP), 6 trichlorophenols (TCP), 6 dichlorophenols (DCP), and 3 mono-chlorophenols (MCP) [3]. CPs are industrially produced by direct chlorination of phenol with chlorine gas, as well as other reac-tions, such as hydrolysis and hydrodechlorination, or by chlori-nation of less chlorinated phenols in the presence of aluminium or iron trichloride [4, 5]. CPs are also by products of industrial processes, such as the bleaching of pulp using chlorine or chlo-rine dioxide [6] and during the production of higher chlorinated phenols [3]. CPs have been detected in groundwater, surface water, waste water, air and soils as a result of their improper dis-posal, leaching from landfills and the incineration of chlorinated wastes [3].
CPs have been identified as priority toxic pollutants by the U.S. Environmental Protection Agency under the Clean Water Act [4, 7-9] due to their environmental persistence, low biode-gradability and potential health hazard. The toxicity of CPs in-creases with the degree of chlorination, but decreases with the degree of their dissociation [4]. As a result, at low pH, where the non-dissociated form of CP is dominant, their toxicity is greater. While PCP has been identified as the most toxic chlorophenol [10], many CPs are of environmental concern due to their acute toxicity and resistance to degradation [1, 2]. In addition to PCP, which has a maximum contaminant level in drinking water of 1 ppb, 2, 4, 6-TCP and 2, 4-DCP are also in the list of drinking water contaminants [11]. CPs have been identified by the Inter-national Agency for Research on Cancer as possible carcinogens [3] and some studies involving the exposure to PCP via inhala-tion and dermal contact have shown the development of specific cancers, such as non-Hodgkin’s lymphoma, multiple myeloma, soft tissue sarcoma and liver cancer [12]. A case study of sawmill workers in New Zealand exposed to PCP reported neuropsycho-logical effects and respiratory diseases in some cases [13]. PCP is also acutely toxic to aquatic microorganisms and fish [10]. Even though the implementation of legislative controls and discharge standards in several countries has curtailed the emissions of CPs from commercial applications, they continue to be identified as common contaminants in surface and groundwater bodies [14]. The environmental prevalence of CPs is attributed to their recalcitrance that results from the carbon-halogen bond, which is cleaved with great difficulty, and the stability of the aromatic structure. Some environmentally significant physical-chemical properties of CPs are presented in Table 1 [15].
Several methods have been developed for treating CP-con-taminated media, including sonication, ozonation [16-18], elec-trochemical treatment [6, 19], advanced chemical oxidation by Fenton’s reagent [4, 20], advanced oxidation [4], bioremediation [18, 21-23], phytodegradation [24], reductive dechlorination by zero valent metals [5, 25-31] and sorption by materials, such as almond shell residues [32], coal fly ash [33], and biosorbents [34, 35]. Furthermore, combinations of treatments have also been used to enhance CP degradation. Examples of such approaches include nanoscale zero valent iron coupled with microwave en-ergy [36] or ultrasound [37], biodegradation following chemical oxidation pre-treatment [38], sequencing chemically reactive media (palladium coated iron) with biological reactive media (sand seeded with anaerobic bacteria) [39] and simultaneous anaerobic-aerobic biodegradation [40].
Of the various treatment approaches, the treatment of CP contaminated groundwater using zero valent metals (ZVMs) is particularly attractive due to its low cost coupled with high ef-ficiency and passive mode of treatment. Accordingly, CP degra-dation by various ZVMs, including iron, zinc, magnesium, and aluminium, has previously been investigated [5, 25, 26, 29, 30, 41]. CP degradation is dependent on the choice of an appropri-ate metal [42]. Table 2 lists the standard electrode reduction po-tentials of various metals in aqueous solution at 25℃ [43, 44]. The electrode potential is the difference between the charge on an electrode and the charge in the solution [44], and metals with higher reduction potentials have a greater ability for degrading chlorinated contaminants in water. However, concerns over the environmental impact and decrease in surface reactivity may limit the use of some promising metals. For example, despite the high reduction potential of zero valent zinc (ZVZn) (-0.763 V), its use has been less than zero valent iron (ZVI) due to the con-cern over water contamination by Zn2+ ions and the decreased ZVZn reactivity by passivation due to the rapid formation of zinc oxides on its reaction with oxygen [45]. The use of zero valent aluminium (ZVAl) for the oxidative degradation of CPs is limited to acidic conditions due to the insolubility of aluminium oxide (Al2O3) layers within the 4 to 9 pH range. Furthermore, concerns over Al3+ ion toxicity to aquatic systems and humans limit the application of ZVAl for the treatment of CPs. In addition, when ZVMs are used in aqueous media, a balance between the metal reactivity with contaminant and with water needs to be main-tained. For example, Sn and Zn are preferable to Mg for treating chlorinated carbons, such as CCl4, as their reactivity with water is lower, but they maintain sufficient reactivity with the contami-nant; Mg oxidation by water overcomes its high reactivity with the contaminant [42].
Among the different ZVMs, ZVI stands out due to its rela-tively high reactivity with halogenated compounds, low instal-lation and operation costs and low environmental impact [26, 28]. Accordingly, ZVI is commonly used as a reactive medium in permeable reactive barriers (PRBs) [46-48], as a cost-effective passive treatment approach often used for
[Table 1.] Selected physical-chemical properties of chlorophenols at 25℃ [15]
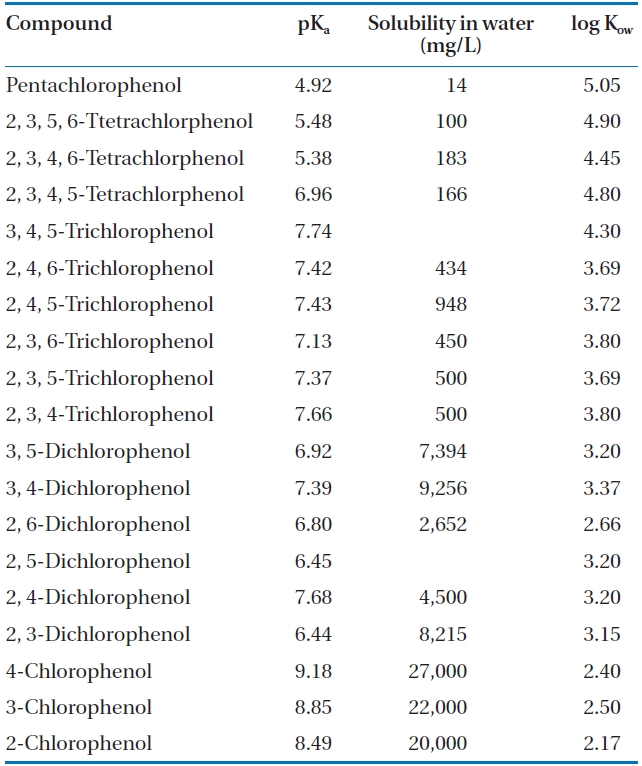
Selected physical-chemical properties of chlorophenols at 25℃ [15]
[Table 2.] Selected standard electrode reduction potential values in aqueous solution at 25℃ [43 44]
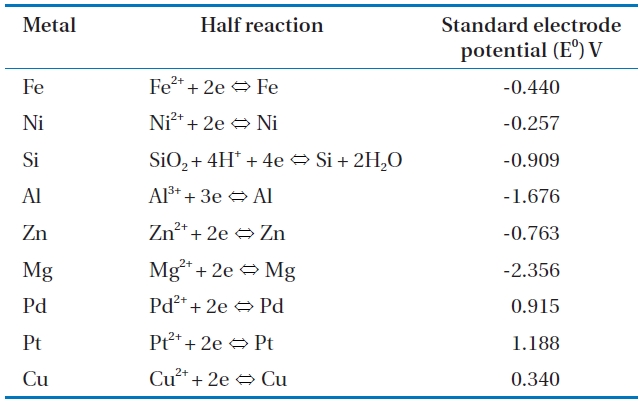
Selected standard electrode reduction potential values in aqueous solution at 25℃ [43 44]
2. Use of ZVI for Reductive Dechlorination of Chlorophenols
While ZVI of all sizes have been used over the past decade to effectively dechlorinate many different chlorinated organic compounds [26, 60-64], later research has aimed at enhancing the degradation using smaller sized ZVI, with several studies having been conducted using microscale ZVI [25, 26, 28, 30, 39, 65] and more recently using nanoscale ZVI [31, 52, 53, 66, 67]. A brief summary of the different types of ZVI used is presented in Table 3. The use of a smaller particle size results in a larger sur-face area and; potentially, increased chemical reactivity [67] as the electronic properties of micro- and nano-scale particles can differ from those of larger particles. The physical and chemical properties of micro to nano sized iron particles are influenced by the atoms located on the iron surface rather than those deeper in the bulk iron material, resulting in more active particles [63]. In addition, nano particles, in contrast to micro scale particles, offer a higher density of reactive sites due to their larger specific surface areas [66]. As nano ZVI are able to stay in suspension for a longer duration, they can be injected into deep or stratified contaminated zones [68].
The rate of dechlorination of many chemicals by ZVI has been reported to decrease with lowering of the degree of chlorination [49, 50]; more chlorinated CPs would be expected to dechlori-nate faster [5]. Also, the degradation rate would be expected to increase for larger iron surface area to solution volume ratios [49, 62]. The ZVI surface has been proposed to contain reactive and non-reactive sites [69-73]; an effective reaction requires the contaminant molecule to come into contact with a reactive site. Reactive sites represent locations on the ZVI surface that de-grade the solute by breaking the bonds in the reactant [69, 70, 72], while non-reactive sites are assumed to be largely respon-sible for the adsorption of contaminant molecules onto the ZVI [70], without causing chemical degradation [72]. Exposed graph-ite inclusions have been reported to be responsible for the non-
[Table 3.] Summary of different types of micro scale ZVI used in previous research

Summary of different types of micro scale ZVI used in previous research
reactive sorption of trichloroethene (TCE) and tetrachloroethyl-ene (PCE) onto cast iron [69]. On the basis of available corrosion literature, Gotpagar et al. [73] hypothesized that defects/abnor-malities present on the surface of iron contribute to the reactive sites. Based on the modelling of the sorption of TCE onto reac-tive and non-reactive sites, with degradation only by the reactive sites, the reactive sites were estimated to account for only 2% of the total number of sites on the surface of ZVI [70], while Bi et al. [71] estimated that only about 2% of the TCE sorption occurred at reactive sites. To complicate matters, when iron is in contact with water, the formation of oxide films on the ZVI surface may mask the reactive and non-reactive sites [74]. The formation of an oxide film can impede the transport and degradation of the organic contaminants by ZVI.
2.1. Processes and Mechanisms of Organic Contaminant Removal in Iron-Water Systems
The removal of dissolved contaminants in iron-water sys-tems occurs by the following four mechanisms:
1) Dechlorination: degradation of the contaminant and forma-tion of intermediates;
2) Sorption: accumulation of the contaminant molecules by ad-sorption or complex formation, without their degradation;
3) Precipitation: immobilization of the contaminant due to the formation of insoluble compounds;
4) Co-precipitation: nonspecific removal of the contaminant via entrapment in the matrix of precipitating or recrystallizing iron oxides/hydroxides on the ZVI surface.
Some common reaction pathways for the removal of dis-solved contaminants (RX) in ZVI/water systems based on above mechanisms are listed in Table 4 [74]. The mechanisms of de-chlorination, sorption and co-precipitation are discussed below, but precipitation has been ignored as this mechanism has pri-marily been reported in relation to the removal of heavy metals.

Summary of possible reaction pathways for aqueous phase contaminant (RX) removal in a ZVI/water system [74]
2.1.1. Dechlorination
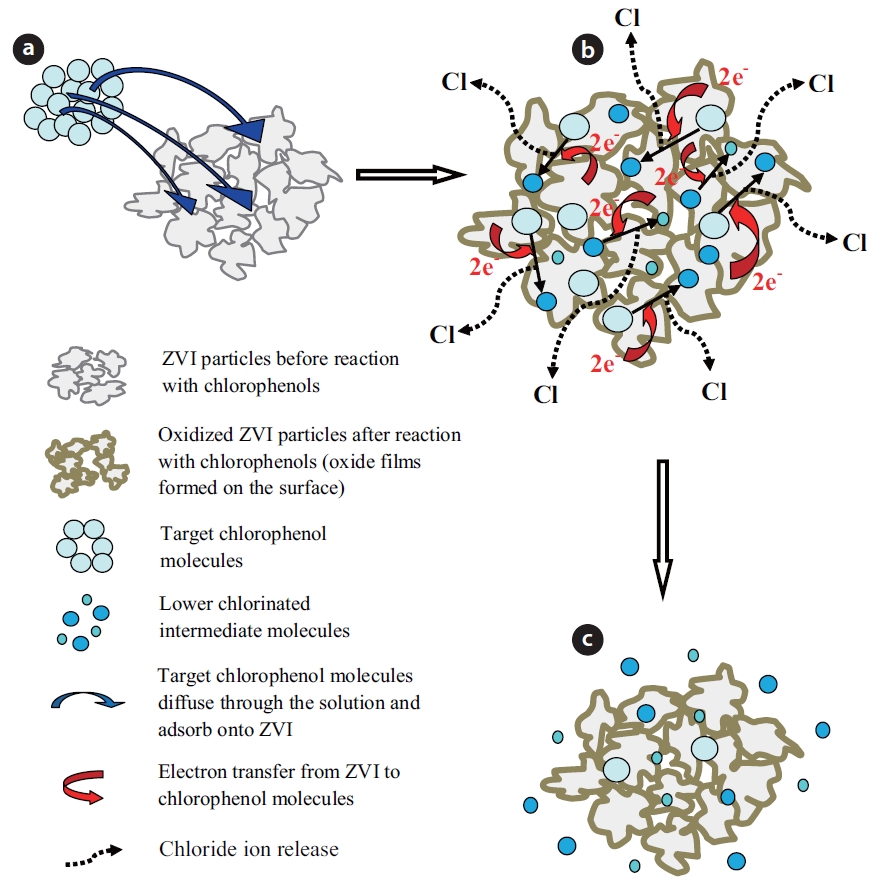
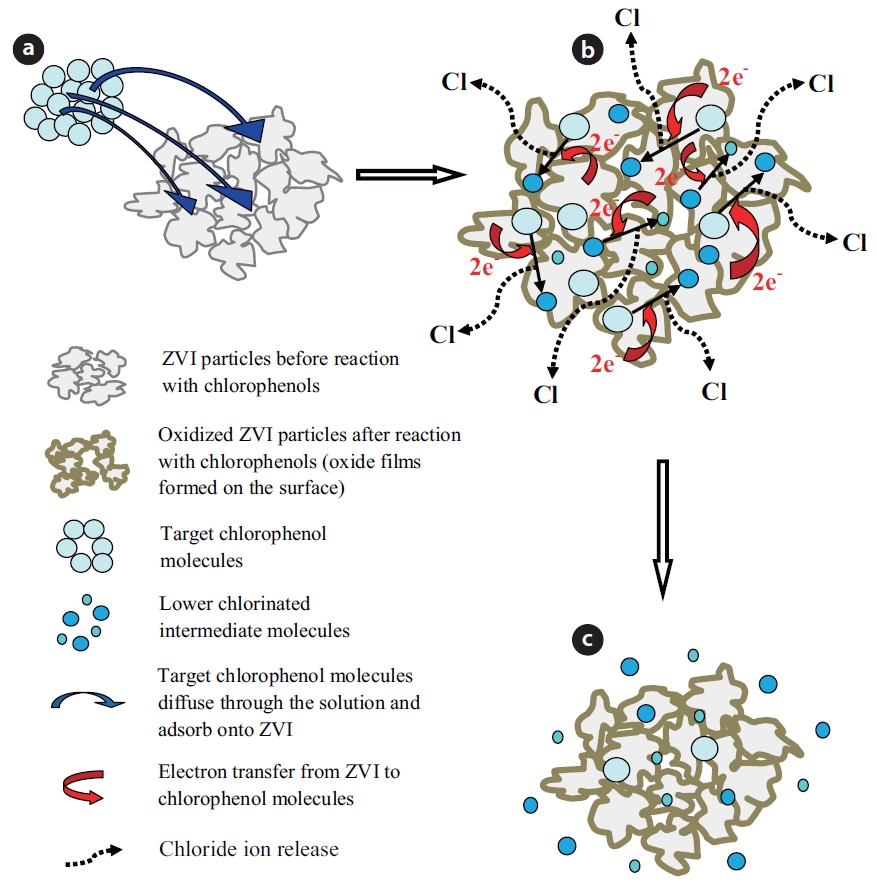
The chemical reduction of chlorinated organic compounds by ZVI-water systems is a surface mediated reaction, which re-sults in direct reduction of the contaminant [47, 50, 75]. Conse-quently, the removal of an organic contaminant by ZVI requires direct contact between the contaminant and reactive sites on the ZVI surface. Several steps are involved during the reaction of a chlorinated contaminant with ZVI [76]. The process of de-chlorination of a contaminant by ZVI is schematically illustrated in Fig. 1.
The contaminant molecules first diffuse through the solution to the ZVI surface, where they adsorb onto the reactive sites (Fig. 1A). Secondly, electrons are transferred from ZVI to the contami-nant molecules and produce lesser chlorinated compounds and various oxides on the surface, with the release of chloride (Fig. 1B). Lastly, some lesser chlorinated products diffuse from the ZVI surface into solution (Fig. 1C). Based on measurements of the large activation energy required for the reaction, Deng et al. [77] concluded that the reduction of vinyl chloride (VC) by ZVI in aqueous systems was controlled by a chemical reaction at the iron surface, instead of the diffusion of VC to the bulk solution. The dechlorination of CPs by ZVI generally follows first order ki-netics [17, 26, 28].
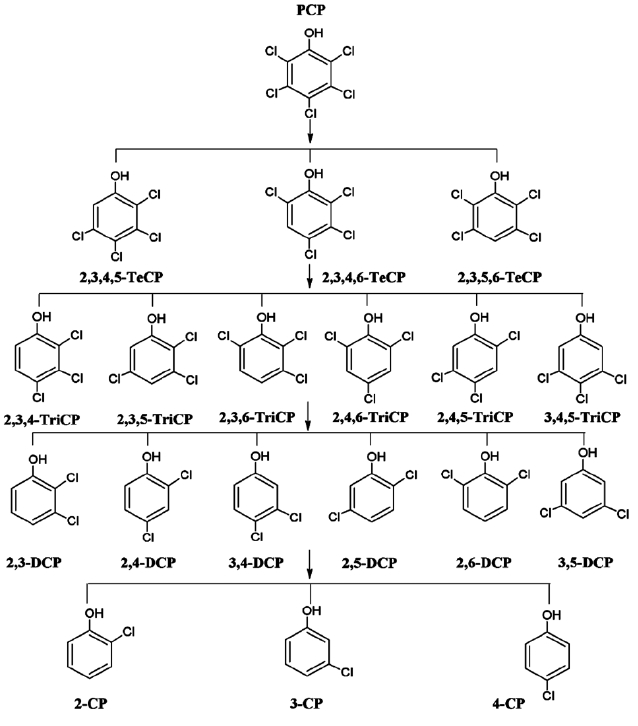
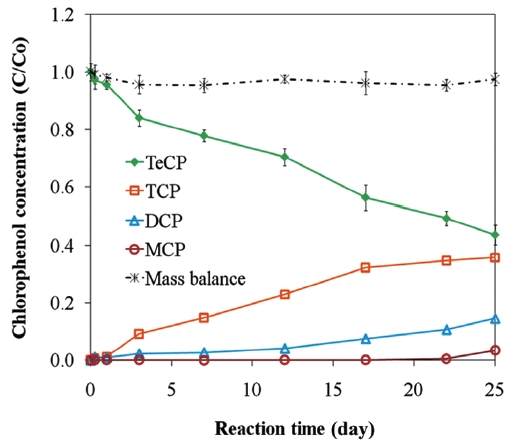
Fig. 2 presents the proposed reaction pathway by which PCP is dechlorinated by ZVI. Data showing the dechlorination of 2, 3, 4, 6-TeCP by ZVI to form TCP, DCP, and MCP is presented in Fig. 3. Equations 1-3 below represent the mechanism for the re-moval of a model chlorinated compound, RCl, via ZVI oxidation coupled with reductive dechlorination of the organic compound
[50]. The anodic reaction for the dissolution of ZVI is illustrated in Equation 1, reductive dechlorination of the chlorinated com-pound by ZVI in the presence of proton donors (for example wa-ter) is presented in Equation 2 and the net reductive dechlorina-tion of the chlorinated compound by ZVI is given in Equation 3.
Fe0 → Fe2+ + 2e- (1)
RCl + 2e- + H+ → RH + Cl- (2)
Fe0 + RCl + H+ → Fe2+ + RH + Cl- (3)
When oxygen is present, the cathodic reduction of oxygen occurs (Equation 4), while ZVI is oxidised to Fe2+ and/or Fe3+ ions, with a rise in the solution pH (Equation 5). Furthermore, under weak acidic to near neutral and oxygenated conditions the Fe2+ ions in water further oxidise to ferric hydroxide; thereby, decreasing the solution pH (Equation 6) [78]. In addition, water can act as an oxidant (Equation 7) and; under anoxic conditions, ZVI corrosion will result in the production of hydrogen and an increase in the solution pH (Equation 8) [50].
O2 + 2H2O + 4e- → 4OH- (4)
2Fe0 + O2 + 2H2O → 2Fe2+ + 4OH- (5)
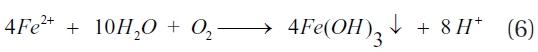
2H2O + 2e- → H2(g) + 2OH- (7)
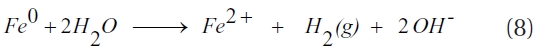
The above reactions lead to an increase in the solution pH, while producing reductants (Fe2+ and H2) with the potential for further dechlorination. However, dechlorination by dissolved Fe2+ occurs slowly and can be further decreased by the presents of ligands in solution due to the formation of complexes with Fe2+ ions [79]. In the absence of a catalyst, H2 is not an effective reductant and its build-up on the metal surface can inhibit the corrosion of ZVI; thereby, slowing the dechlorination reaction [50]. A survey of the literature relating to the reduction of chlori-nated hydrocarbons by iron materials indicated that direct elec-tron transfer as well as an indirect mechanism involving atomic hydrogen participate in dechlorination [80, 81].
Negatively charged chloride ions are released during de-chlorination, which adsorb onto the oxide films and penetrate through the pores/defects on the oxides. This causes dispersion of the oxide films; thereby, increasing their permeability and en-hancing the movement of ions across the oxide films. Chloride ions are essential for the initiation of pitting and crevice corro-sion. Enhanced degradation rates of chlorinated compounds by ZVI may be due to enhanced metal dissolution and electron transfer due to pitting corrosion [82]. Furthermore, the chem-istry of the ZVI surface during the dechlorination reaction is similar to the classical crevice corrosion of steel in the presence of chloride ions [83]. The oxidation of iron releases electrons, which reduces dissolved oxygen to hydroxide ions. In addition, dissolved oxygen depletion in crevices leads to an increase in the
concentration of metal cations in the small crevices, which at-tract and increase the diffusion of negatively charged chloride ions from the bulk solution into the crevices. This process leads to an increase in the rate of metal dissolution [84]. Conversely, during this process, the formation of insoluble metal hydroxides can lead to coating of the exterior surface of ZVI and reduce the rate of metal dissolution [73].
2.1.2. Sorption
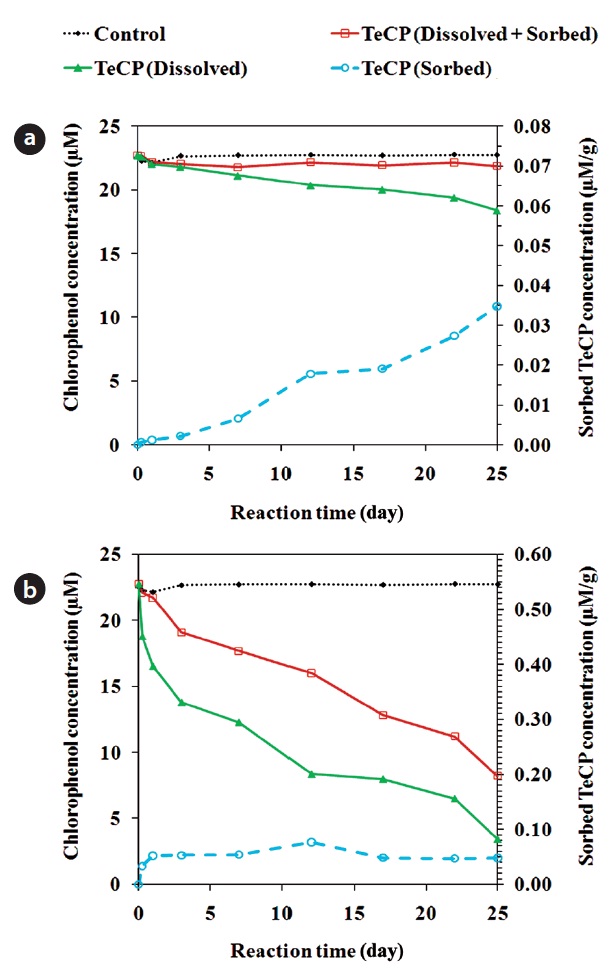
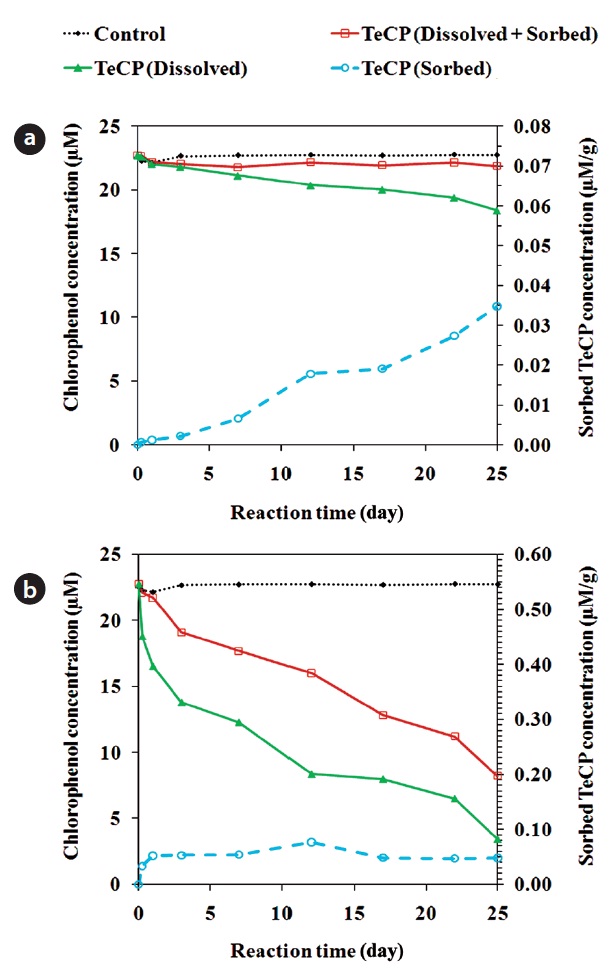
In addition to dechlorination, CPs dissolved in solution can be removed via sorption onto the ZVI surface [26, 85]. Fig. 4 shows the amounts of 2, 3, 4, 6-TeCP sorbed onto unmodified and acid pretrated ZVI. In some cases, the sorption onto ZVI can be sufficiently strong to prevent the diffusion of sorbed re-actant molecules back into solution. In such cases, the sorbed molecules can hinder the access of dissolved CPs to recative sites on the ZVI surface, attenuating the degradation process. The sorption onto ZVI may be a significant removal mechanism, with some studies having reported the removal of over 50% of the initial PCP mass via this process [26]. TCE and PCE sorb pri-marily onto the non-reactive sites of ZVI [72]. The adsorption of CPs onto iron oxides occurs via the phenolate group [86]. A high solution pH leads to decreased surface reactivity and enhanced adsorption of TeCPs onto the ZVI surface [87]. Under moder-ately acidic conditions (pH around 5.4), the adsorption of CPs is influenced by their degree of dissociation; CPs with lower pKa values have lower solubilities in water and show greater tenden-cies to adsorb onto iron oxides [87]. Contaminants with greater hydrophobicity are more susceptible to sorption, irrespective of their reactivity with ZVMs [25]. The adsorption of CPs dissolved in water to iron oxides is less than that of those in the gaseous phase, suggesting that hydrophobic CPs have a modest capac-ity to compete with water for adsorption onto hydrophilic oxide surface sites [86].
2.1.3. Co-precipitation
Co-precipitation refers to contaminant entrapment in the iron oxides/hydroxides precipitating or recrystallizing on the ZVI surface. The co-precipitation of contaminants during iron corrosion is a fundamental mechanism of dissolved contami-nant removal by ZVI [88]. At pH values within the range 4 to 9, ZVI corrosion results in the formation of an oxide film on its sur-face. At lower pH values, the oxide films are relatively porous, unstable and detachable, while at higher pH values, the oxide films are more stable. At neutral pH, various iron oxides (green rusts, hydroxides, and oxyhydroxides) can precipitate, poten-tially entrapping dissolved organic compounds on the ZVI sur-face. In contrast to earlier studies, which considered oxide films as inhibitory to contaminant removal [50, 75], co-precipitation can lead to the oxide films acting as contaminant scavengers; contaminant removal may not necessarily be limited to surface mediated processes [89].
2.2. Challenges to Implementation of ZVI Technology
While ZVI is effective at treating CPs dissolved in water, it does not effectively degrade dense non-aqueous phase liquids (DNAPLs) in a source area. Because the ZVI surface is hydrophil-ic, while CPs are typically hydrophobic, the interaction between ZVI and separate phase CPs tends to be absent [63, 90]. Further-more, the rate of reduction is greatly influenced by surface char-acteristics of ZVI, such as the morphology, presence of impuri-ties, specific surface area and crystallinity [76]. One of the major challenges in the use of ZVI is the decrease in the iron reactivity over time, which has been attributed to:
1) Changes in the iron surface morphology [73];
2) Surface passivation due to saturation of the reactive sites with metal oxides or other species, depending on the solution composition, and the generation and accumulation of lesser chlorinated by products [82];
3) Precipitation of metal hydroxides (e.g., Fe(OH)2 and Fe(OH)3) and metal carbonates (e.g., FeCO3) on the iron surface [62, 65].
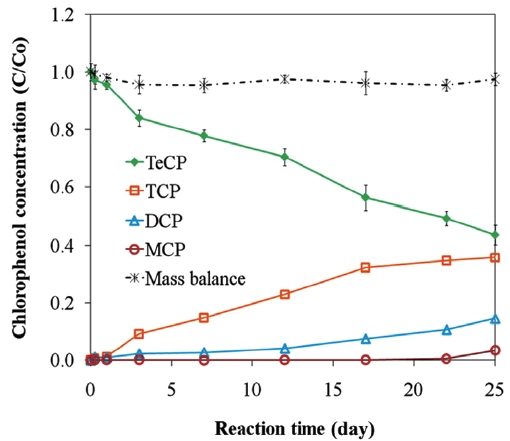
Low rates of dechlorination by ZVI have also been partially attributed to the excessive sorption of contaminants to nonreac-tive sites and the impeding of electron transfer by oxide layers [73]. However, the reactivity of the ZVI surface can be sustained in the presence of some oxides, such as magnetite (Fe3O4) and green rust (Fe [II]/Fe [III] hydroxide), while the presence of goe-thite (α-FeOOH) and hematite (α-Fe2O3) decrease the reactivity [91-93]. The formation and role of iron oxides in the removal of CPs is further discussed in Section 4. The inhibition of iron cor-rosion and reduction reactions can also result from excessive H2 accumulation on the iron surface [50]. Furthermore, the deg-radation of chlorinated aromatic compounds and compounds with lesser degrees of chlorination by ZVI occurs at slower rates
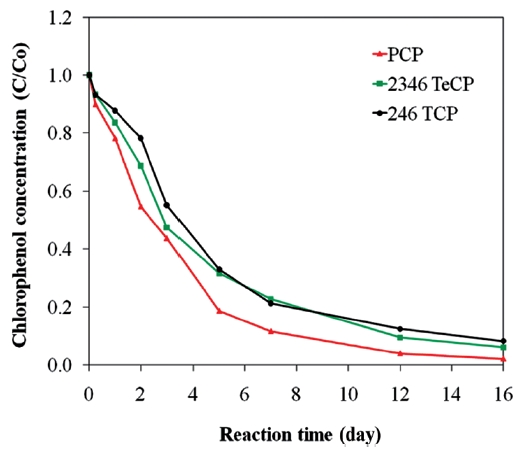
[56, 65]. This sequence is illustrated for the degradations of PCP, 2, 3, 4, 6-TeCP, and 2, 4, 6-TCP in Fig. 5. The accumulation of chlorinated by products due to low ZVI reactivity with lesser chlorinated contaminants can give rise to toxicological concern. Lastly, competition with co-contaminants, such as nitrate and chromate, can inhibit or decrease the reduction of chlorinated compounds [61, 94].
The use of nano ZVI in real applications is hindered by the rapid agglomeration of nano particles to micro scale particles and the larger aggregates in water [67] and the propensity of nano ZVI particles to interact with the subsurface media [68]. These processes may affect the mobility and reactivity of the nano particles, limiting their capacity to dechlorinate the con-taminants. An approach proposed to mitigate this issue involves enhancing the stability of the nano iron particles using natural clay minerals and zeolite [95]. However, in comparison to micro scale iron particles the lifespan of nano iron particles is expected to be smaller due to the relatively faster background corrosion [96].
Bimetallic systems have been investigated as a means of over-coming the limitations associated with the unmodified ZVI used to degrade chlorinated organics [25, 26, 28, 37, 54, 97-99]. These systems are a combination of two metals - a primary metal and a secondary metal, selected based on their standard reduction po-tentials (Table 2); the secondary metal is electropositive relative to the primary metal. In bimetallic systems, the primary metal, such as ZVI, is known to serve as the electron donor, while the secondary metal (Pd, platinum (Pt) or Ni) acts as a catalyst [45]. During the reaction, the secondary metal is not consumed, but increases the rate and efficiency of the reaction [45, 58, 100-102]. The incorporation of the catalyst contributes to increase the sur-face area and number of reactive sites on the ZVI surface [58, 100]. Catalysts, such as Pd and Ni, have a void orbit, which allows for the formation of complex compounds by combining with the chloride atoms of chlorinated molecules; thus, reducing the ac-tivation energy and increasing the dechlorination reaction rate, while limiting the production of chlorinated intermediate com-
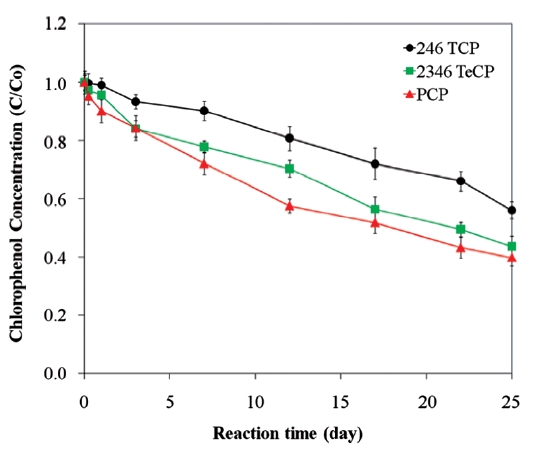
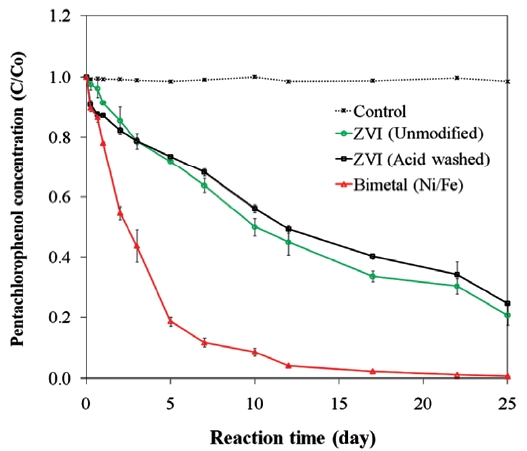
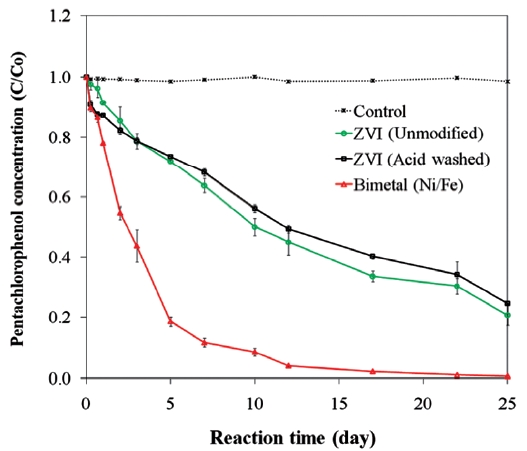
pounds [45, 99]. The presence of a secondary metal may also provide a surface that is less prone to fouling and that can con-tinue to supply electrons for the complete oxidation-reduction process [73]. In the presence of a catalytic metal, dechlorination of chlorinated organic compounds occurs via hydrogenation [96]. The catalytic reactivity of bimetals and the dechlorination rate depend on the properties of the chlorinated contaminants and the ZVI [26, 56]. For example, while Kim and Carraway [26] observed that bimetallic systems were poorer at dechlorinating PCP than unmodified iron, Fig. 6 presents data that show ZVI bi-metals can lead to faster PCP dechlorination than unmodified or acid washed ZVI. Similarly, differences in molecular structures cause isomers of chlorophenols to show different dechlorination rates upon reacting with bimetals [56].
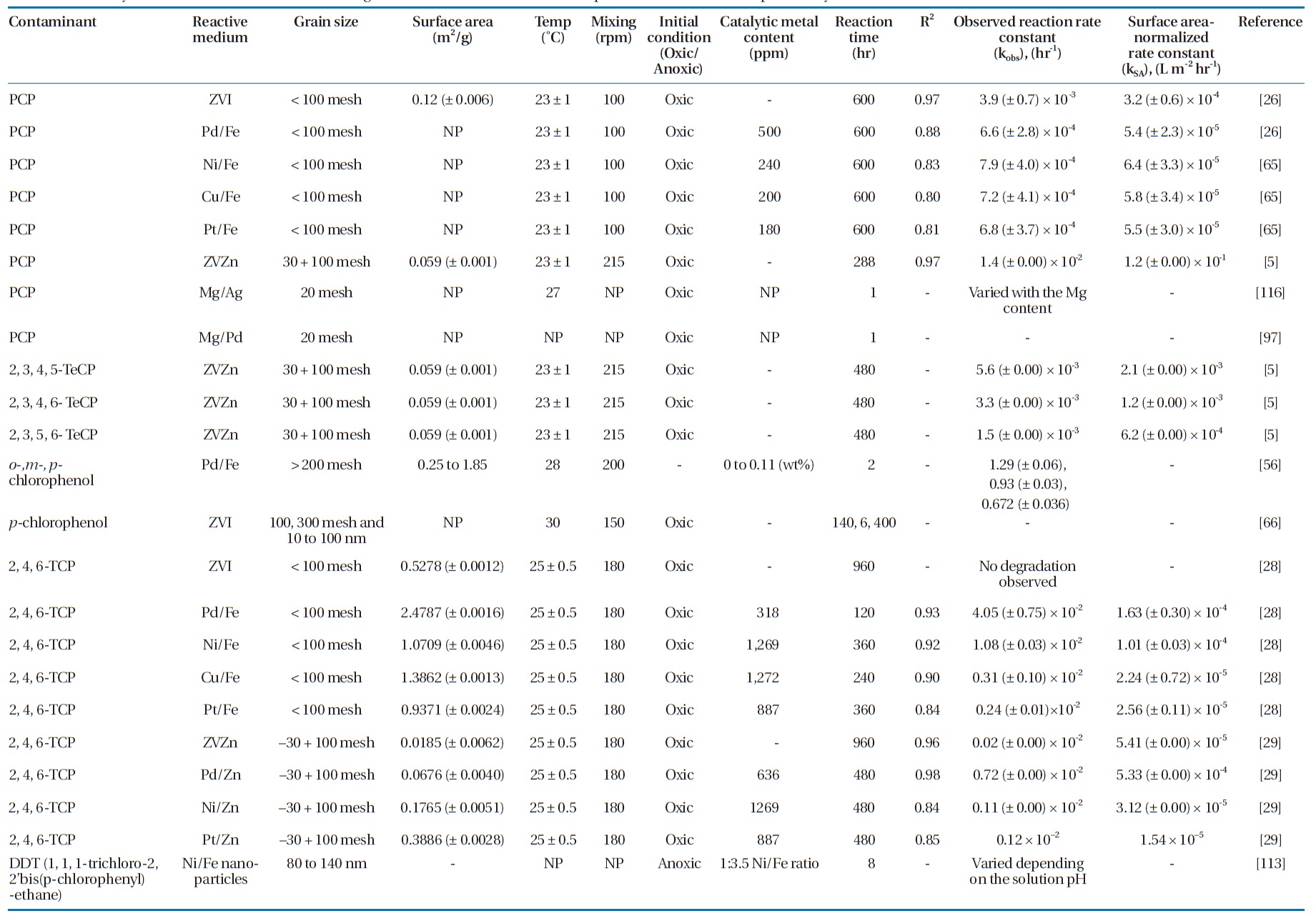
Several bimetallic combinations have been studied for their efficiencies to degrade chlorinated organic compounds, includ-ing ZVI/Pd (Fe/Pd), ZVI/Ni (Fe/Ni), ZVI/Copper (Fe/Cu), and Magnesium /Palladium (Mg/Pd) [63]. Table 5 summarizes the degradation rate constants for selected chlorinated aliphatics and aromatics by different metallic media. Increasing the cata-lytic metal loading on the iron surface has been observed to in-crease the dechlorination efficiency [37, 78, 103]. This effect was attributed to the increased availability of the catalyst, where a higher catalytic metal load results in greater dispersion of the catalyst on the ZVI surface as well as increases in the surface area and number of dechlorination sites [56, 103].
3.1. Different Methods of Bimetallic Systems
Simple bimetallic systems prepared by physically mixing the primary metal (for example ZVI) and the catalyst (Ni) at pre-determined weight ratios have been shown to display superior performance [30, 31, 45, 96, 104, 105]. For example, a mixture of nano-Ni and nano-ZVI showed greater PCP dechlorination than nano-ZVI particles alone [31]. However, bimetallics are usually prepared via reductive deposition by doping a catalytic metal, such as Pd, Pt or Ni (which are not active by themselves), onto the primary reactive metal, such as ZVI or ZVZn [25, 26, 28, 53]. The reductive adsorption of the catalytic metal onto the iron sur-face is depicted in Equations 9 [78] and 10 [54]:
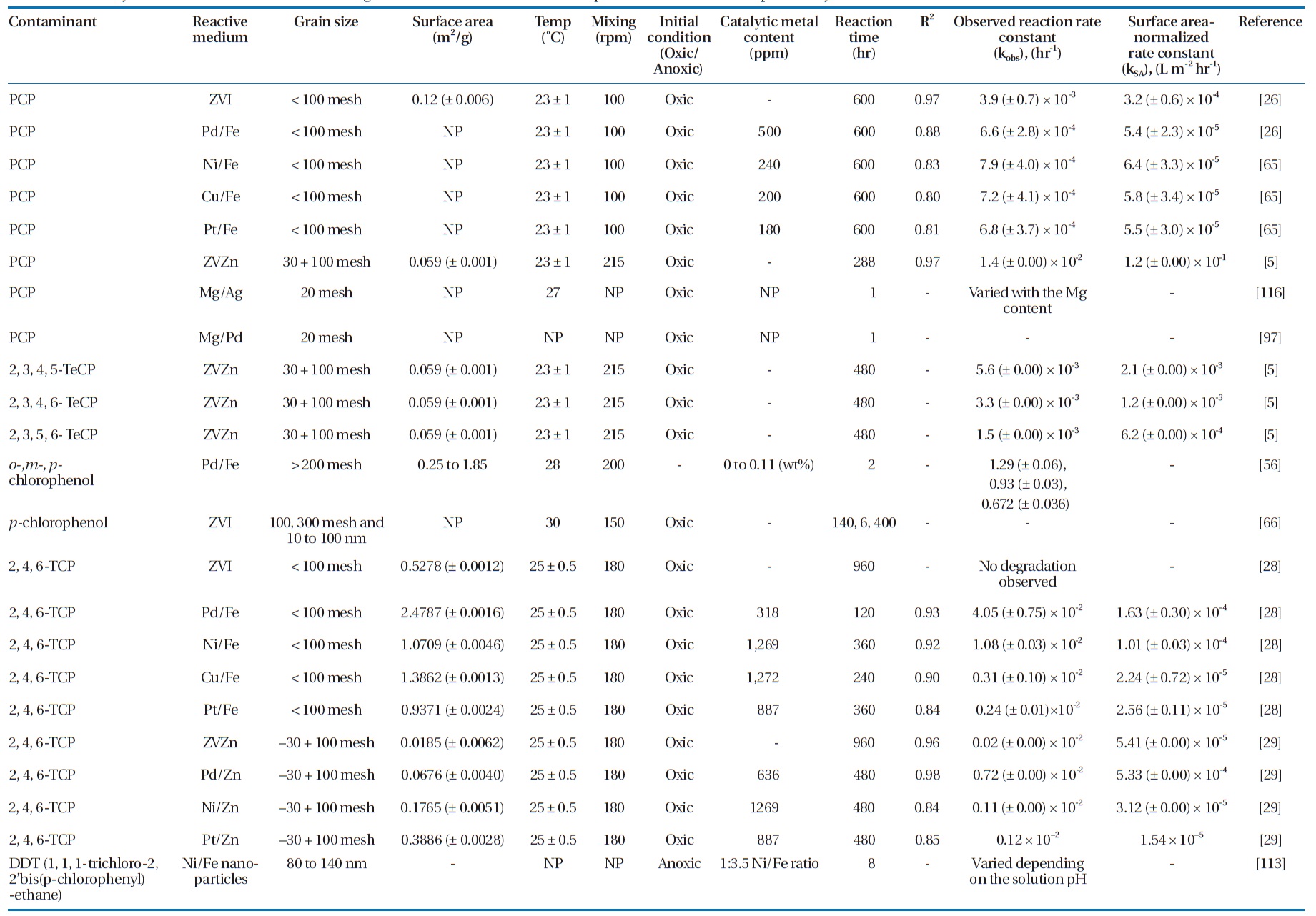
Summary of reaction rate constants in the degradation of several chlorinated aliphatic and aromatic compounds by various metals/metal combinations
Pd4+ + 2Fe0 → Pd0 + 2Fe2+ (9)
2Fe0 + Ni2+ → Ni0 + 2Fe2+ (10)
A bimetallic system can contain several galvanic cells on the primary metal; thus, increasing the rate of corrosion of the iron, which leads to an increased rate of dechlorination [52]. However, some studies have reported that physical mixing of two metals did not result in an increased rate of degradation of chlorinated aromatic compounds [45]. Comparisons of the reactivities of physical mixtures of nano-Ni and nano-ZVI with nano-ZVI alone and with Ni coated nano-ZVI showed that the Ni coated nano-ZVI gave the highest dechlorination activity [96].
3.2. Processes and Mechanisms of Organic Contaminant Removal in Bimetallic Systems
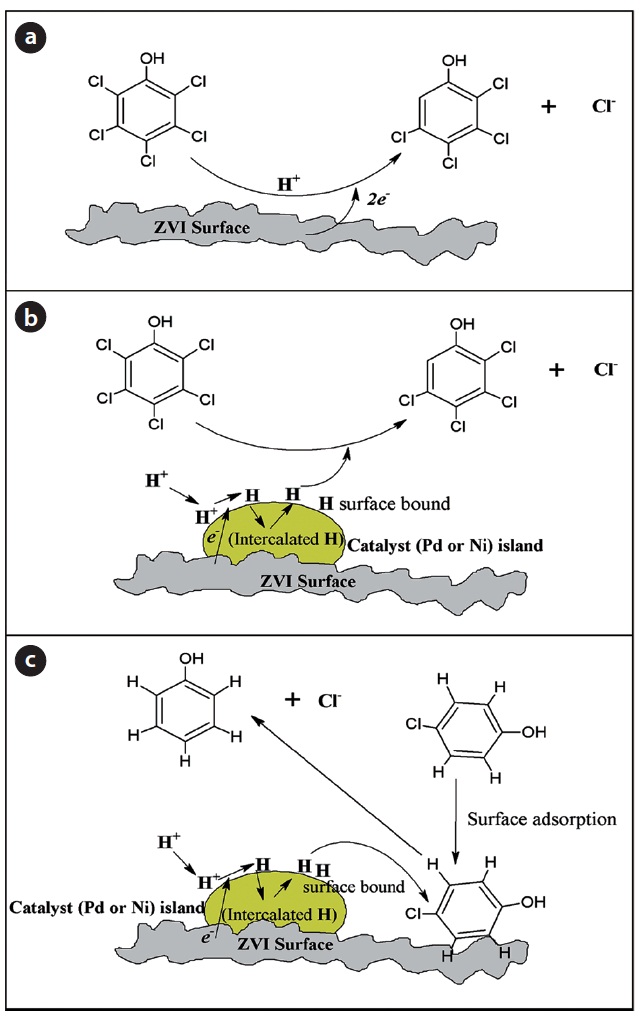
Several mechanisms have been proposed to explain the im-proved dechlorination rates observed with bimetallic systems. Fig. 7 presents the various mechanisms for the hydrodechlori-nation of CPs by bimetallic particles [29, 106]. Fig. 7A illustrates the dechlorination via direct electron transfer from ZVI to the chlorinated molecule. A common hypothesis attributes the en-hanced reactivity of bimetals to the formation of galvanic cou-ples between the ZVI and catalyst due to the flow of electrons from ZVI to the catalyst driven by the difference in their elec-trode potentials. In the case of bimetals containing iron, ZVI acts as the anode and the catalyst as the cathode [45, 84, 96, 100, 107, 108]. Although a higher rate of ZVI corrosion would be expected in the presence of a catalyst, with a weaker reductant, possessing a positive reduction potential compared to ZVI, the dissolutions of ZVI and Ni would be significantly lower in the Ni/Fe bimetal compared to the individual metals [96]; the presence of ZVI has been reported to inhibit Ni corrosion [96, 108].
Another hypothesis attributes hydrogen adsorption onto the catalyst surface for the enhanced dechlorination by the bimet-als. This hypothesis assumes that chlorinated organic molecules are adsorbed onto the catalyst surface, where reduction of the chlorinated organic compounds occurs on the surface of the catalytic metal, but not at the interface of the catalyst and ZVI (Fig. 7B) [96, 106, 109-111]. In a bimetallic system (
2ZVIC + H2 → 2ZVIC? Hads (11)
ZVIC + H2O + e- → ZVIC ?Hads + OH- (12)
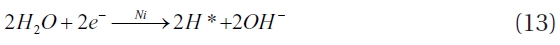
Ni + H* → Ni ? H (14)
Ni ? H + R ? Cl → Ni + R ? H + Cl- (15)
A three step process has been proposed for the dechlorina-tion that occurs within bimetallic systems [99]. First, hydrogen gas is produced from the corrosion of ZVI in water, while the sec-ond step involves the transport and adsorption of chlorinated contaminant molecules onto the catalyst surfaces via bonding between the chlorine and the catalyst surface. The last step is de-chlorination, which involves the incorporation of hydrogen into the contaminant molecule and the release of chloride ions into solution. Another hypothesis paralleling that suggested above is that contaminant molecules adsorbed onto the ZVI surface are dechlorinated at the catalyst/ZVI interface (Fig. 7C) [106].
Furthermore, some studies have postulated that rather than the atomic hydrogen adsorbed on the reductant surface, the atomic hydrogen absorbed/intercalated within the lattice of the catalytic metal is the dominant reductant responsible for the re-ductive dechlorination of the chlorinated compounds adsorbed onto the catalyst surface [52, 106, 110, 114]. The enhanced re-activity of the bimetallic particles has been attributed to the adsorption of chlorinated contaminant and the generation and
storage of reactive hydrogen molecules on the metal surface. Under this hypothesis, the metal with the lower potential in the bimetal will preferentially dissolve and release electrons (e.g., Zn dissolution will occur in the Pd/Zn pair), while the other metal, with the relatively higher (positive) reduction potential, will act as the catalyst [73]. A consensus on the most likely hypothesis for the enhanced dechlorination by bimetals is currently lack-ing;clear experimental studies examining the above hypotheses are limited, and doping of ZVI with catalytic metals has been seen to not always result in an enhanced rate of degradation. For example, lowering of the PCP and 1, 1, 1-trichloroethane reduc-tion rates reportedly occurred when ZVI was modified with Pd, Pt, Ni or Cu [26].
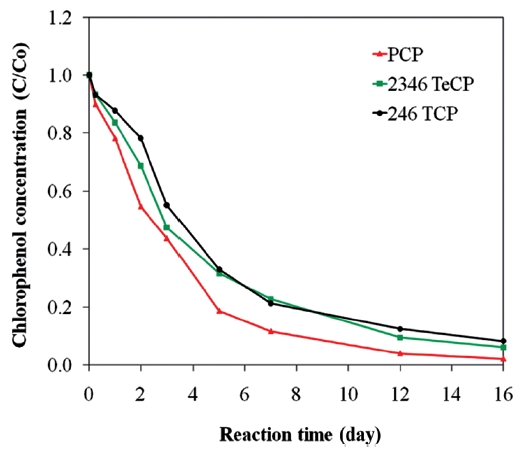
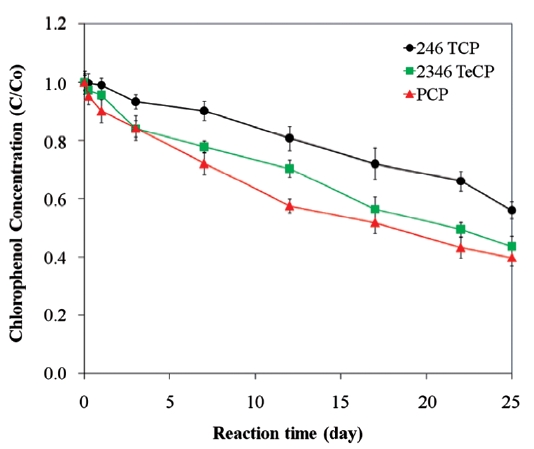
The dechlorination of CPs by bimetallics follows first order kinetics [28, 54-57, 59], and the rate constants have shown to be dependent on the degree of chlorination of the organic com-pound. A hypothesis explaining this behaviour is that the energy required to break the C-Cl bond differs for penta- tetra-, tri-, di-and mono-chlorophenols. In bimetallic systems, the rate and extent of degradation decreases with higher degrees of chlorina-tion, i.e., the order for the rates of degradation is MCP > DCP >TCP > PCP [25, 97, 115]. The more chlorinated compounds expe-rience greater repulsion between electrons on the metal surface and the negatively charged chlorine of the chlorinated organic molecule, leading to greater sorption of the lesser chlorinated organics [81]. As a result, in bimetallic systems, where indirect reduction through atomic hydrogen predominates, more chlo-rinated compounds may exhibit lower degradation rates com-pared to the less chlorinated compounds [81]. There is another hypothesis for the increased rate of degradation on lowering of the dechlorination, which is based on the assumption that dechlorination occurs in two steps. The first step is the cleav-age of the C-Cl bond resulting in the formation of a transient electron-deficient carbocation on the phenol ring; the second is the bonding of the carbocation with an electrophile, such as a hydrogen from the metal hydrides formed on the iron surface, to complete its octet configuration [97]. A greater number of chlorines on the phenol ring intensify the positive charge arising from C-Cl cleavage, which destabilizes the carbocation; there-by, lowering the rate of dechlorination [97]. However, contrary to the above observation, other studies have shown the inverse trend with lesser chlorinated compounds degrading at lower rates [101, 116]. Fig. 8 presents data illustrating such an effect for the degradations of PCP, TeCP, and TCP using a Ni/Fe bimetal, where the rate and extent of PCP degradation was higher com-pared to those of TeCP and TCP. Therefore, the only consistent conclusion is that the efficiency and rate of hydrodechlorination of CPs by bimetallic systems are dependent on certain factors, such as the type and characteristics of the metal combinations used and the characteristics of the target contaminant.
3.3. Advantages of Using Bimetals
Bimetals can efficiently catalyse dechlorination reactions and reduce the amount of toxic chlorinated by products [56, 78, 96], and further progress dechlorination reactions towards completion [45]. The dominant reaction for the dechlorination of chlorinated compounds in bimetallic systems is via hydrogen reduction rather than the electron transfer process from ZVI at the solid/liquid interface [45, 56, 78, 102]. Furthermore, the ad-dition of a catalytic metal renders stability to the iron particles by lowering the oxidation of ZVI and formation of iron oxides on the surface [107]. While Fe/Pd particles were not observed to undergo a colour change upon exposure to air, ZVI particles rap-idly underwent surface oxidation, turning from black to reddish-brown within a few hours [45]. Similarly, ZVI particles lost their reactivity within a few days, while the activity of Fe/Pd particles was maintained beyond two weeks [52] and Pd doping eliminat-ed the passivation of the Zn surface [45].
3.4. Limitations of Using Bimetallic Particles
Bimetallic surfaces can lose their reactivity over time through disintegration of the catalyst due to ZVI dissolution, the forma-tion of a hydrogen gas layer on the bimetallic surface due to excessive hydrogen production in the system, the formation of iron oxides on the surface [59], and the sorption of chlorinated organic compounds [117]. During dechlorination, the formation of ferrous and ferric hydroxides on the iron surface can passivate the bimetallic surface by hindering the transport of chlorinated molecules as well as blocking access to reactive sites on its sur-face [62]. Additionally, the rate of dechlorination can decrease over time due to the formation of hydrates and oxides on the bimetal surfaces [37, 59, 96]. Some investigators have argued that the formation of iron oxide, occlusion of organic molecules and accumulation of hydrogen are temporary, reversible effects, which can be concealed by controlling the solution pH under anoxic conditions [59, 117].
4. Formation, Characterisation and Role of the Iron Oxide Films
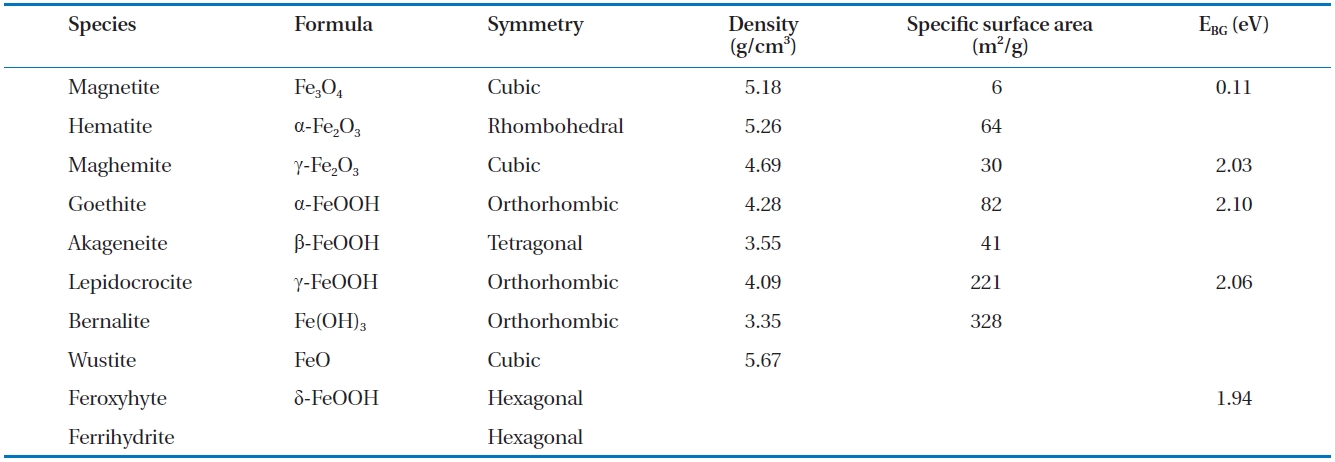
The development of iron oxide films occurs naturally when ZVI comes in to contact with air and water. Likewise, when groundwater flows through a reactive barrier filled with ZVI or a bimetal, iron corrosion and other chemical reactions can lead to the formations of different iron oxide films on the ZVI surface [89]. The type and characteristics of the iron oxide films formed depend on the environmental conditions, such as groundwater chemistry, solution pH and availability of dissolved oxygen. As an example, in a ZVI/water system maintained at near neutral pH, oxides, such as magnetite and hematite, hydroxides, such as ferrous and ferric hydroxide, and oxyhydroxides, such as goe-thite and green rusts, are formed [74]. The properties of the ox-ide films, such as their electronic conductivity and porosity, can influence the corrosion of iron in natural waters [118], and the formation of some oxides can enhance or decrease the reactivity of the iron surface [119]. The formation of oxides can also lower the porosity of the reactive medium in PRBs; considering the pH range 4 to 9 in natural waters, the oxides formed at a low pH are relatively porous (i.e., unprotective) and removable compared to the more stable and protective oxides formed at higher pH val-ues [120]. Numerous oxides can form on ZVI, including magne-tite, green rust (chloride and sulphate), lepidocrocite, goethite,maghemite, akageneite, wustite and ferrihydrite [31, 91, 94, 119, 121-129]. Table 6 provides a summary of selected characteristics of some of these oxides [130-132].
The characteristics of iron oxides can affect the effectiveness of the catalytic metal deposited onto the ZVI surface. Further-more, iron oxides can sorb dissolved organic compounds. Ha-logenated hydrocarbons usually sorb via hydrophobic bonding rather than complex formation with iron oxides. Chlorinated organic molecules containing other functional groups can strongly sorb via interactions between the reactive sites on ox-ides and the functional groups [133]. Such interactions depend on the affinity of the contaminant molecules for the sorption sites, shape and polarity of the molecule, and the location of the functional group [133]. Furthermore, dissolved organics can also be removed from solution via co-precipitation, a process in which contaminants initially adsorb onto fresh iron oxides, but are later entrapped within the oxide as it matures [74].
Iron oxide films play several different roles [47, 94, 119, 122, 126, 134-137]. Oxide precipitates can passivate the iron surface by creating a barrier between the contaminant molecules and the ZVI surface, hindering the electron transfer process and suppressing dechlorination [122]. Passivation of iron surfaces occurs via the formation of oxides/oxyhydroxides, such as he-matite, maghemite, lepidocrocite and goethite [94, 135, 138]. The passivating oxide films are multilayered, nanocrytalline microstructures, typically consisting of an inner magnetite layer and an outer maghemite or hematite layer [91, 122, 135, 139]. Conversely, some iron oxides can act as semiconductive films by contributing electrons from their conduction band to the dechlorination reaction; thereby, promoting contaminant re-
[Table 6.] Selected characteristics of iron oxides
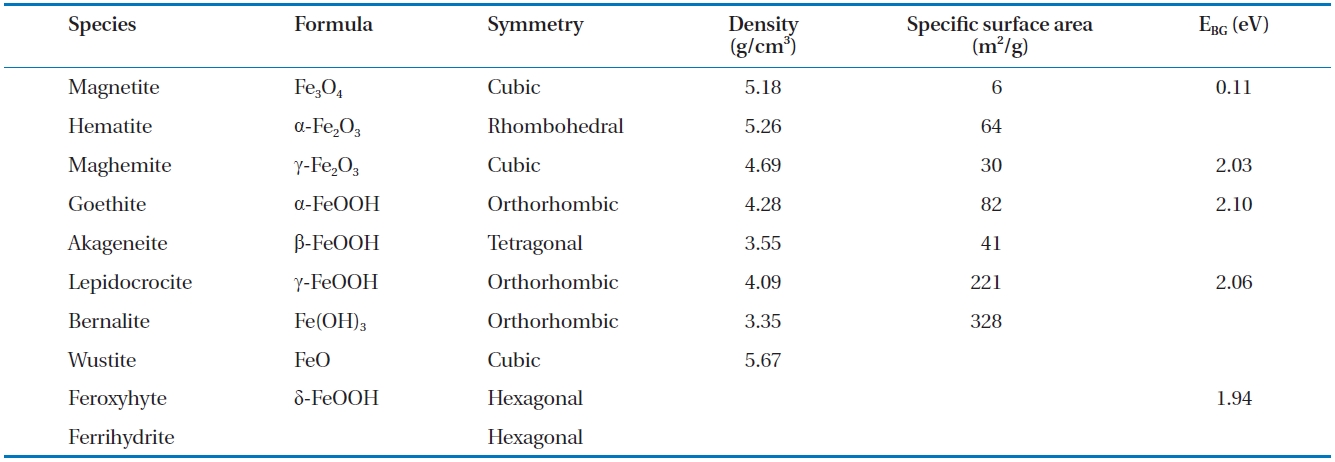
Selected characteristics of iron oxides
duction [122]. Oxides, such as magnetite and green rust, allow electron transfer and facilitate the dechlorination reaction [94, 119, 135, 138]. However, green rusts are unstable and will trans-form over time to relatively more stable oxides, such as goethite, lepidocrocite, maghemite or magnetite, depending on certain factors, such as the solution pH, and rates of oxidation and de-hydroxylation [132, 140-142]. Similarly, while magnetite is elec-trically conductive, its formation can lead to ZVI passivation if it hinders the release of Fe2+ to the bulk solution [91]. Conversely, precipitation of the passivating iron oxides can in some cases (e.g., when it occurs on non-reactive sites of the ZVI surface) re-sult in little or no decrease in the reactivity of ZVI [70]. In addi-tion to the commonly accepted mechanisms for the dechlorina-tion of organic compounds - direct electron transfer from ZVI or indirect electron transfer through the oxide layer, iron oxides can act as coordinating surfaces to reduce contaminants using the electrons contributed by surface bound Fe2+ or by surface reac-tive sites of Fe2+ ions formed in oxide films (structural Fe2+) due to the release of electrons [122, 143].
In recent years, the effect of specific oxides on the reactiv-ity with chlorinated organic compounds has been investigated using pure iron oxides [143-146]. Magnetite degradation of PCP showed the reduction to be controlled by surface reaction mechanisms, such as sorption and oxidation [145], and that the reactivity decreased with an increase in the particle size of mag-netite [144]. The rate of carbon tetrachloride dechlorination by magnetite increased on increasing the solution pH from 6 to 10 [143]. Nonetheless, the presence of co-contaminants affects the reactivity of iron oxides [134, 145]. Different species present in a matrix (e.g., anions, ligands, other organic compounds) com-pete to adsorb onto the reactive sites available on the oxide sur-faces [145], where strong adsorption of such co-contaminants could inhibit the dechlorination of the target compounds [134].
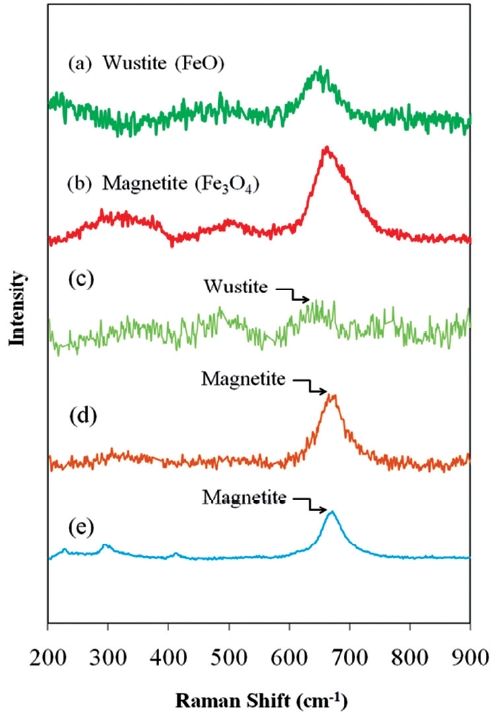
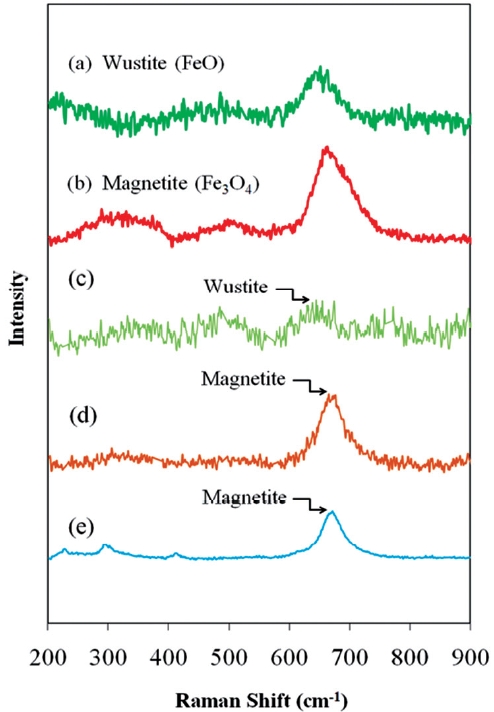
Fig. 9 presents the Raman spectra of typical iron oxides de-tected on ZVI/bimetallic surfaces. The unwashed ZVI surface contained Wustite, while acid washing and Ni coating during the bimetal preparation resulted in increased proportions of mag-netite present on the respective surfaces. The higher reactivity of acid washed ZVI and Ni/Fe was partially attributed to the in-creased proportion of magnetite.
5. Effect of Environmental Parameters and Matrix Characteristics
The solution pH is a critical environmental parameter af-fecting the degradation of CPs. The solubility of CPs in water generally increases at higher pH values [2, 14] and the solution pH affects the rates of iron corrosion and oxide formation. Iron dissolves under acidic conditions, while under basic conditions iron oxides form and cause surface passivation [50, 62]. The de-chlorination efficiency decreases with increasing solution pH [27, 37, 55, 97, 112, 113]. Moreover, with bimetallic systems at basic pH conditions, less atomic hydrogen or metal hydride par-ticipate in the dechlorination reaction. Therefore, extremes of solution pH can influence the availability of reactive sites on the ZVI and bimetals for dechlorination.
Dissolved oxygen (DO) is another significant parameter af-fecting the dechlorination of CPs by ZVI and bimetals. The pres-ence of DO can hamper the dechlorination by ZVI and bimetals because oxygen is the preferred electron acceptor under oxic conditions [50]. In an oxic system, DO can compete with CPs for the electrons produced via ZVI oxidation [147]. Furthermore, the presence of DO can affect the formation and composition of the oxide films formed on ZVI/bimetallic surfaces [135]. Under oxic conditions, the rates of dechlorination tend to be significantly lower to those under anoxic conditions [112, 136, 148].
The composition of contaminated groundwater can affect the efficiency of a ZVI or bimetallic system for treating CPs. Groundwater can contain several constituents that may po-tentially compete with CPs for electrons, such as natural or-ganic matter [149, 150] and dissolved inorganic anions, such as nitrate, chloride, chromate, carbonate, phosphate, sulphate, and sulphide [124, 126, 151, 152]. The presence of dissolved co-contaminants can affect the corrosion of ZVI and the interaction between CPs and ZVI/bimetals. Furthermore, the behaviour of CPs when present as an individual component may differ from that when present as a multi-component system. Anions, such as nitrate and chromate, can compete for reactive sites and de-crease the sorption capacity and reactivity of the ZVI/bimetallic surfaces due to the formation of passive iron oxides on the sur-face [61, 94, 153, 154].
CPs are a group of industrially important chemicals. Howev-er, due to their unique characteristics (e.g., aromaticity stability, hydrophobic nature, toxicity to humans and ecosystems and en-vironmental persistence), the treatment of CPs has been a chal-lenge. The use of ZVI is an emerging passive

![Selected physical-chemical properties of chlorophenols at 25℃ [15]](http://oak.go.kr/repository/journal/11631/E1HGBK_2011_v16n4_187_t001.jpg)
![Selected standard electrode reduction potential values in aqueous solution at 25℃ [43 44]](http://oak.go.kr/repository/journal/11631/E1HGBK_2011_v16n4_187_t002.jpg)

![Summary of possible reaction pathways for aqueous phase contaminant (RX) removal in a ZVI/water system [74]](http://oak.go.kr/repository/journal/11631/E1HGBK_2011_v16n4_187_t004.jpg)
![Mechanisms for the hydrodechlorination of chlorophenols by bimetallic systems. ZVI: zero valent iron [29 106].](http://oak.go.kr/repository/journal/11631/E1HGBK_2011_v16n4_187_f007.jpg)