Inflammatory mediators such as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) have been implicated in various inflammatory diseases. In this study, we investigated the anti-inflammatory activities of Chondria crassicaulis ethanolic extract (CCE) by measuring its effects on the expression of iNOS and COX-2 proteins in lipopolysaccharide (LPS)-treated RAW 264.7 murine macrophages. CCE significantly and dose-dependently inhibited the LPS-induced release of nitric oxide and pros-taglandin E2, and suppressed the expression of iNOS and COX-2 proteins in LPS-stimulated RAW 264.7 cells, without causing any cytotoxicity. It also inhibited the production of the pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α in LPS-stimulated RAW 264.7 cells. Moreover, treatment with CCE strongly suppressed nuclear factor-κB (NF-κB) promoter-driven expression in LPS-treated RAW 264.7 cells. CCE treatment blocked nuclear translocation of the p65 subunit of NF-κB by preventing proteolytic degradation of inhibitor of κB-α. These results indicate that CCE regulates iNOS and COX-2 expression through NF-κB-dependent transcriptional control, and identifies potential candidates for the treatment or prevention of inflammatory diseases.
Macrophages are major inflammatory cells and immune effector cells. Activation of macrophages occurs in inflamed tissues and is induced by exposure to interferon-γ, tumor ne-crosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and micro-bial lipopolysaccharide (LPS) (Xie et al., 1993). Activated macrophages produce excessive amounts of inflammatory mediators such as nitric oxide (NO) and prostaglandins (PGs), in addition to various pro-inflammatory cytokines (Nathan, 1992; Zhang and Ghosh, 2000). Excessive produc-tion of inflammatory mediators and cytokines contributes to the pathogenesis of chronic diseases such as atherosclerosis, inflammatory arthritis, and cancer (Libby, 2006; Packard and Libby, 2008; Solinas et al., 2010). Substances that inhibit the production of these molecules are considered to be potential anti-inflammatory agents.
The expression of inflammatory proteins such as cyclo-oxyganase-2 (COX-2) and inducible nitric oxide synthase (iNOS) and pro-inflammatory cytokines is primarily con-trolled at the transcription level (D’Acquisto et al., 1997; Kim and Moudgil, 2008). Transcriptional induction of COX-2 and iNOS is largely dependent on the cooperative activity of multiple transcription factors, including nuclear factor-κB (NF-κB) and activator protein 1 (AP-1), which act on cog-nate
Marine algae have been identified as rich sources of structurally diverse bioactive compounds with great phar-maceutical potential (Abad et al., 2008; Blunt et al., 2010). A variety of biological compounds, including phlorotannins and fucoxanthin, were isolated from marine algae and char-acterized in terms of their biological activities (Kim et al., 2005, 2009, 2011; Woo et al., 2009). In Korea,
Dried powder from
>
Cell culture and viability assay
RAW 264.7 murine macrophages (ATCC, Rockville, MD, USA) were cultured at 37℃ in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA) supple-mented with 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin sulfate (100 μg/mL) in a humidified atmo-sphere containing 5% CO2. Cell viability was determined by a 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay using a CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (Pro-mega, Madison, WI, USA) according to the manufacturer’s manual. Briefly, cells were inoculated at a density of 1 × 105 cells/well into 96-well plates and cultured at 37℃ for 24 h. The culture medium was replaced with 200 μL of serial dilu-tions of CCE (0-200 μg/mL), and the cells were incubated for 24 h. The culture medium was removed and replaced by 95 μL of fresh culture medium plus 5 μL of MTS solution. After 1 h, the absorbance at 490 nm was measured using an Ultraspec 2100 Pro microplate reader (GE Healthcare Bio-Sciences Co., Piscataway, NJ, USA).
RAW 264.7 cells (1 × 106 cells/mL) were transferred to 12-well plates, incubated with 0-200 μg/mL CCE for 1 h, and stimulated with 1 μg/mL LPS for 24 h. Cell culture medium was collected after centrifugation at 2,000 g for 10 min. The nitrite concentration in the culture medium was measured as an indicator of NO production. Then, 100 μL of the culture supernatant was mixed with the same volume of Griess re-agent (0.1% naphtylethylene diamine dihydrochloride and 1% sulfanilamide in 5% H3PO4). The absorbance of the resulting mixture at 540 nm was measured with an Ultraspec 2100 Pro microplate reader. The concentration of nitrite was calculated using sodium nitrite as a standard.
>
Enzyme-linked immunosorbent assay (ELISA)
ELISA was used to assess the inhibitory effect of CCE on the production of PGE2 and the cytokines TNF-α, IL-1β, and IL-6 in LPS-stimulated RAW 264.7 cells. RAW 264.7 cells (1 × 106 cells/mL) were transferred to 12-well plates, treated with 0-200 μg/mL CCE for 1 h, and stimulated with LPS (1 μg/mL) for 24 h. After incubation, the supernatants were collected and assayed with the appropriate ELISA kit (R&D Systems, Min-neapolis, MN, USA) according to the manufacturer’s protocol.
RAW 264.7 cells were washed twice with ice-cold phos-phate-buffered saline (PBS), lysed through incubation in buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Non-idet-40, 1% Tween 20, 0.1% sodium dodecyl sulfate, 10 μg/mL leupeptin, 50 mM NaF, 1 mM PMSF) on ice for 30 min. After centrifugation at 18,000 g for 10 min, the protein con-tent of the supernatant was measured. Protein aliquots (40 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (10%) and transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). The membranes were washed with Tris-buffered saline (10 mM Tris, 150 mM NaCl) supplemented with 0.05% (v/v) Tween 20 (TBST) and then blocked with TBST containing 5% nonfat dried milk (w/v). The membranes were incubated at 4℃ with antibod-ies specific for COX-2 (dilution 1:2,000; incubation period 10 h), iNOS (1:2,000; 10 h), p-IκB-α (1:2,000; 15 h), IκB-α (1:2,000; 15 h), NF-κB (1:2,000; 15 h), and actin (1:5,000; 10 h). The membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. The membranes were washed three times with TBST at room temperature. Immunoreactivity was detected using ECL reagent (GE Healthcare Bio-Sciences Co.). Equal protein loading was assessed by measuring the actin protein levels.
>
Transient transfection and luciferase assay
One microgram of murine NF-κB promoter/luciferase DNA (Stratagene, Santa Clara, CA, USA) was transiently transfect-ed with 200 ng of control pRL-TK DNA (Promega) into RAW 264.7 cells (2 × 106 cells/well) in a 24-well plate using Lipo-fectamine/Plus reagent (Invitrogen, Carlsbad, CA, USA) for 24 h. Cells pretreated with 0-200 μg/mL CCE were stimulated with LPS (μg/mL) for 6 h. Each well was then washed twice with cold PBS and harvested in 100 μL of lysis buffer (0.5 mM HEPES [pH 7.8], 1% Triton N-101, 1 mM CaCl2 1 mM MgCl2) and analyzed for luciferase activity using a lucifer-ase assay kit. Luminescence was measured using a TopCount microplate scintillation and luminescence counter in single-photon counting mode (0.1 min/well) following adaptation for 5 min in the dark. Luciferase activity was normalized to the expression of pRL-TK.
>
Preparation of cytosolic and nuclear extracts
RAW 264.7 cells were treated with various concentrations of CCE for 1 h and stimulated with LPS (1 μg/mL) for 30 min. Cells were washed twice with cold PBS, scraped into 200 μL of cold PBS, and pelleted through centrifugation of 300 g for 5 min. Cell pellets were resuspended in hypotonic buffer
(10 mM HEPES/KOH, 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, pH 7.9) and incubated on ice for 15 min. After vortexing for 10 s, homogenates were centrifuged at 13,000 g for 10 min. The resulting supernatant (cytosolic extract) was collected. The pellet was gently resus-pended in 20 μL of complete lysis buffer (50 mM HEPES/KOH, 50 mM KCl, 1 mM DTT, 300 mM NaCl, 0.1 mM EDTA, 10% glycerol, 0.5 mM PMSF, pH 7.9) and centrifuged at 13,000 g for 20 min at 4℃. The resulting supernatant was used as a nuclear extract.
Data were expressed as the mean ± SD. Data were ana-lyzed by one-way analysis of variance (ANOVA), and pairs of groups were then analyzed by Student’s
>
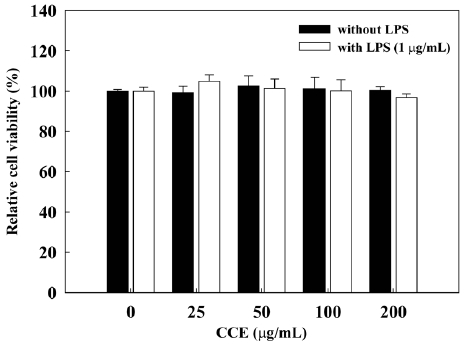
Effect of CCE on RAW 264.7 cell viability
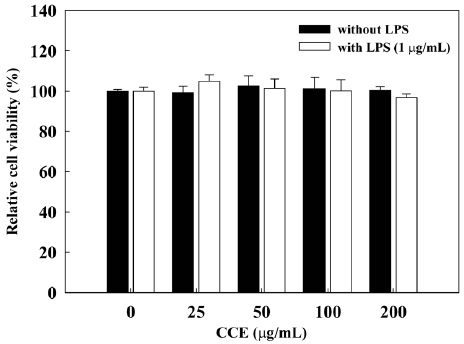
To determine whether CCE causes cytotoxicity in RAW 264.7 cells, we assessed the effects of various concentrations of CCE on RAW 264.7 cell viability by MTS assay (Fig. 1). We found that CCE, alone or combined with LPS (1 μg/mL), did not affect the viability of RAW 264.7 cells at concentra-tions of up to 200 μg/mL. At a concentration of 400 μg/mL, CCE reduced the viability of RAW 264.7 cells to 78.8 ± 4.8% when applied alone and to 82.5 ± 5.1% when applied with LPS. Thus, we used CCE at concentrations of ≤200 μg/mL, which did not affect cell viability, in further studies of anti-inflammatory activity and possible mechanisms of action in RAW 264.7 cells.
The results of several studies strongly suggest that phenolic compounds derived from botanical sources are able to sup-press inflammation via removal of reactive oxygen species by antioxidants (Rahman et al., 2006; Kim and Kim, 2010). We previously described the anti-oxidative and anti-inflammatory activities of phloroglucinol derivatives of
>
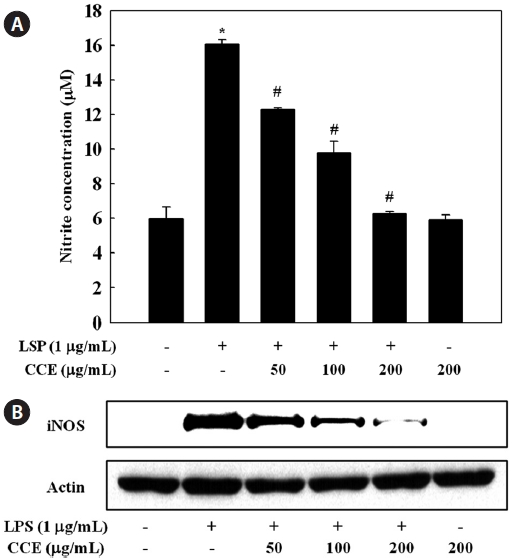
Effect of CCE on the production of NO in LPS-stimulated cells
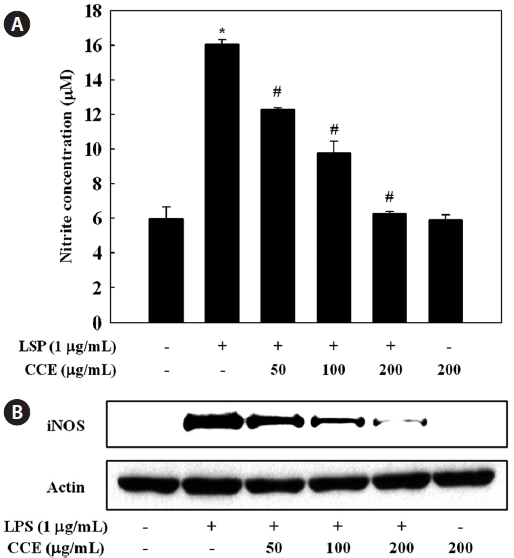
To determine the effect of CCE on NO production in LPS-stimulated RAW 264.7 cells, we measured culture medium ni-trite concentrations using the Griess reagent. RAW 264.7 cells were pretreated with various concentration of CCE (50-200 μg/mL) for 1 h and stimulated with LPS for 24 h. Production of NO, measured as nitrite, was increased by LPS. CCE sig-nificantly and dose-dependently suppressed the production of NO by LPS-stimulated cells (
NO is synthesized from ?-arginine and molecular oxygen in a reaction catalyzed by NOS. Under pathological conditions, a significant increase in iNOS-derived NO contributes to the induction of inflammation, acting synergistically with other inflammatory mediators (Nathan, 1992). iNOS is strongly induced following exposure to bacterial endotoxin and pro-inflammatory cytokines (Guha and Mackman, 2001). Com-pounds able to reduce NO production or iNOS activity may be attractive anti-inflammatory agents, and for this reason, the suppressive effects of natural marine compounds on NO pro-duction have been intensively studied with the aim of devel-oping anti-inflammatory drugs (Abad et al., 2008; Jung et al., 2009; Heo et al., 2010; Jin et al., 2010; Kim and Kim, 2010). The present results suggest that CCE-mediated inhibition of NO production in LPS-stimulated macrophages was associ-ated with downregulation of the iNOS protein.
>
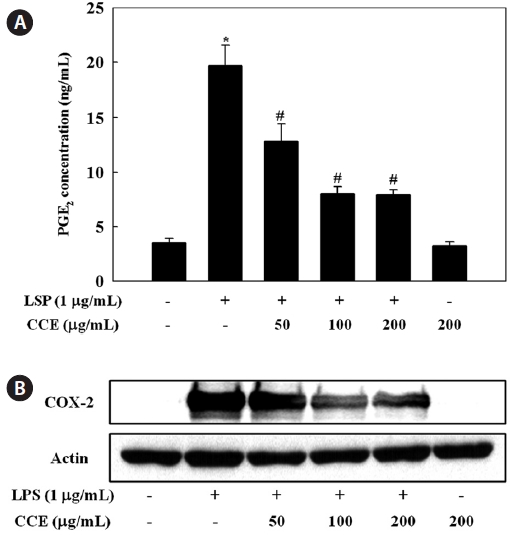
Effect of CCE on PGE2 production in LPS-stimulated cells
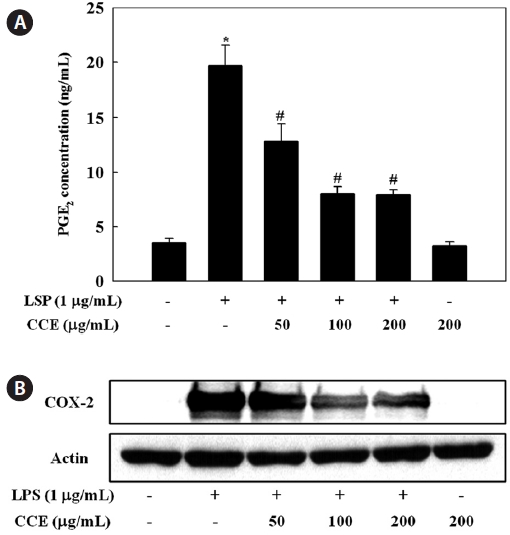
To evaluate the effect of CCE on PGE2 production in LPS-stimulated RAW 264.7 cells, culture medium PGE2 concentra-tions were determined by ELISA. The stimulation of PGE2 production in RAW 264.7 cells by LPS was suppressed by CCE treatment (
COXs regulate the conversion of arachidonic acid to PGE2 and are rate-limiting enzymes in the biosynthesis of PGs (Du-
bois et al., 1998). COX-1 is constitutively expressed in many tissues, while COX-2 is known as an inducible enzyme that, in most cases, generates large amounts of PGs. COX-2 is highly expressed in inflammation-related cell types including macro-phages and mast cells stimulated with pro-inflammatory cy-tokines and/or LPS (Nathan, 1992; Vane et al., 1994). Recent studies have shown that
>
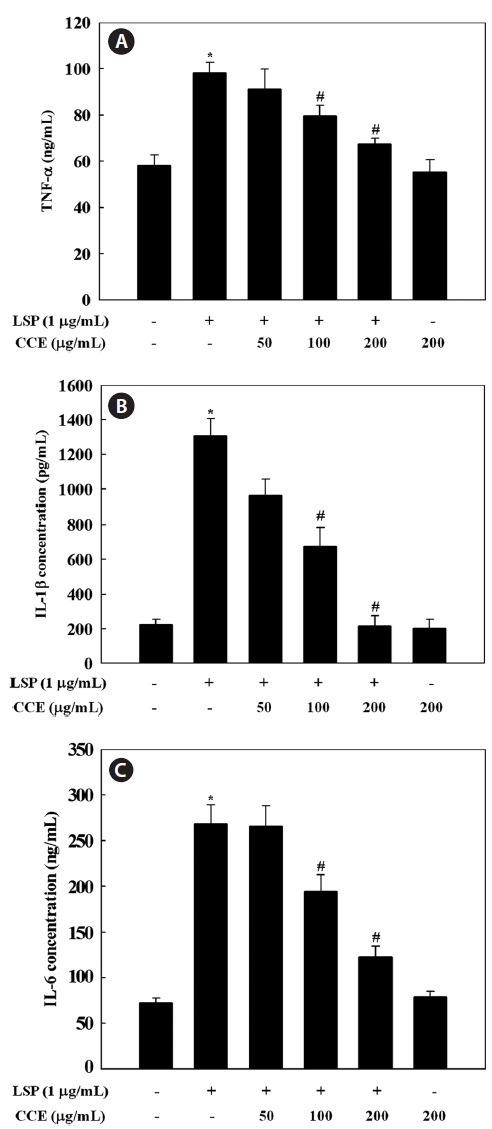
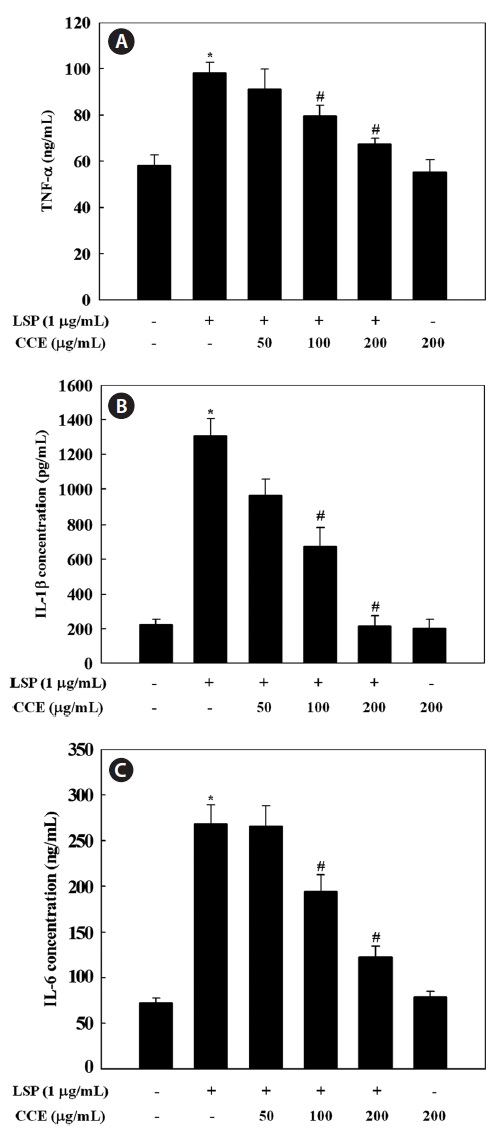
Effect of CCE on the production of TNF-α, IL-1β, and IL-6 in LPS-stimulated cells
Since CCE was found to inhibit the inflammatory mediators NO and PGE2 in a dose-dependent manner, we investigated the inhibitory effect of CCE on LPS-induced TNF-α, IL-1β,
and IL-6 release by ELISA. Stimulation of RAW 264.7 cells with LPS significantly increased the levels of TNF-α, IL-1β, and IL-6. However, CCE, at concentrations ranging from 50 to 200 μg/mL, dose-dependently inhibited cytokine production in LPS-stimulated RAW 264.7 cells (
inhibinhibitory effect of CCE on TNF-α, IL-1β, and IL-6 produc-tion was not attributable to cytotoxic effects, since cell viabil-ity was not altered by CCE at the concentrations used in this study (Fig. 1).
Pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 are small secreted proteins that regulate immunity and inflammation. Bacterial LPS stimulates macrophages to re-lease TNF-α. Secreted TNF-α or LPS then induces cells to release IL-1β and IL-6 (Beutler and Cerami, 1989). TNF-α induces several physiological processes, including septic shock, inflammation, and cytotoxicity (Dinarello, 1999). IL-1β is a major pro-inflammatory cytokine, which is mainly re-leased by macrophages and is believed to play a key role in the pathophysiology of endometriosis (Lebovic et al., 2000). IL-1β is also important in the initiation and enhancement of inflammatory responses to microbial infection (Kim and Moudgil, 2008). IL-6 is a pivotal pro-inflammatory cytokine that is mainly synthesized by macrophages. It plays a role in the acute-phase immune response (Yoshimura, 2006) and is regarded as an endogenous mediator of LPS-induced fever. The results of the present study suggest that CCE significantly suppressed LPS-induced TNF-α, IL-1β, and IL-6 secretion, which supports the idea that CCE inhibits the initial phase of a LPS-stimulated inflammatory response.
>
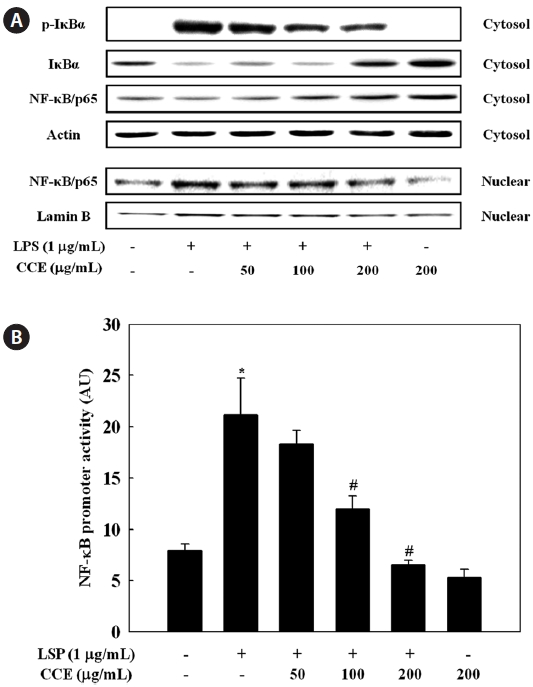
Effect of CCE on NF-κB activation in LPS-stimulat-ed cells
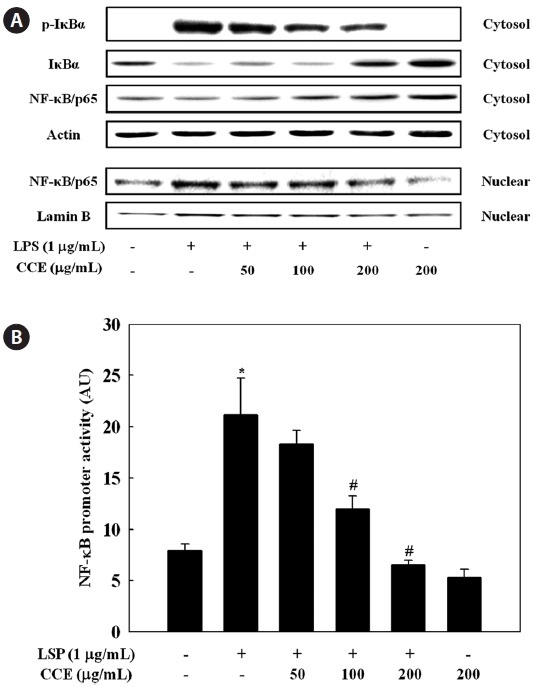
We further investigated whether CCE could inhibit the LPS-stimulated degradation of IκB-α in RAW 264.7 cells by Western blotting. LPS treatment resulted in degradation of IκB-α, a response that was dose-dependently reversed by CCE pretreatment (Fig. 5A). To investigate translocation of the p65 subunit of NF-κB from the cytosol to the nucleus after its release from IκBs, we examined NF-κB levels in cytosolic and nuclear extracts. CCE pretreatment dose-dependently increased cytosolic NF-κB levels (Fig. 5A). CCE dose-de-pendently reduced the nuclear p65 level, and in parallel in-creased the cytosolic p65 level. Considering the inhibitory effects of CCE on LPS-induced NF-κB activation, we next determined the effect of CCE on NF-κB promoter activity in LPS-stimulated macrophage cells. To achieve this, cells were transiently transfected with a luciferase construct containing the murine NF-κB promoter, pretreated for 2 h with various concentrations of CCE, and treated with LPS (1 μg/mL) for 6 h. Luciferase assay data show that CCE treatment significantly suppressed LPS-induced NF-κB promoter-driven luciferase expression in macrophages (
The induction of inflammatory mediators such as NO and PGE2, and of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6, is dependent on NF-κB activation (Li and Ver-
ma, 2002). NF-κB is known to play a pivotal role in the regu-lation of cell survival genes and to coordinate the expression of pro-inflammatory enzymes and cytokines such as iNOS, COX-2, TNF-α, IL-1β, and IL-6 (Xie et al., 1993; D’Acquisto et al., 1997; Marks-Konczalik et al., 1998; Makarov, 2001). NF-κB is associated with and tightly controlled by an inhibi-tory subunit, IκB, which retains NF-κB in the cytoplasm in an inactive form. Activation of NF-κB by LPS involves the phosphorylation of IκB-α kinase, which phosphorylates IκB-α at Ser32 and Ser36, leading to the subsequent degradation of IκB-α and inducing translocation of NF-κB into the nucleus (Chen et al., 1995). In the present study, we observed that deg-radation of IκB-α in response to LPS treatment was reversed by CCE treatment, suggesting that CCE protected IκB-α from proteolytic degradation. Moreover, we found that transloca-tion of NF-κB into the nucleus was dose-dependently inhib-ited by CCE. From these data, the CCE-mediated downregula-tion of LPS-induced iNOS, COX-2, TNF-α, IL-1β, and IL-6 expression in RAW 264.7 cells is likely largely dependent on the ability of CCE to inhibit NF-κB signaling. This is, to the best of knowledge, the first report addressing CCE’s negative regulation of LPS-activated NF-κB signaling.
In conclusion, we demonstrated that CCE inhibited pro-duction of the inflammatory mediators NO and PGE2 and the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 in LPS-stimulated RAW 264.7 macrophages. Moreover, the inhibitory effects of CCE were found to be associated with the inactivation of NF-κB signaling through inhibition of IκB degradation. Verification of CCE’s anti-inflammatory activity and mechanisms of action in cells will be beneficial to the fur-ther application of CCE in functional foods in the treatment of inflammatory diseases. These results suggest the need to perform additional studies to identify the compounds in CCE that contribute to its anti-inflammatory activity.