In developing a useful tool to detect parasitic dynamics in an estuarine ecosystem, a denaturing high-performance liquid chromatography (DHPLC) assay was optimized by cloning plasmid DNA from the grass shrimp Palaemonetes pugio, and its two parasites, the trematode Microphallus turgidus and bopyrid isopod Probopyrus pandalicola. The optimal separation condition was an oven temperature of 57.9℃ and 62-68% of buffer B gradient at a flow rate of 0.45 mL/min. A peptide nucleic acid blocking probe was designed to clamp the amplification of the host gene, which increased the amplification efficiency of genes with low copy numbers. Using the DHPLC assay with wild-type genomic, the assay could detect GC Gram positive bacteria and the bopyrid isopod (P. pandalicola). Therefore, the DHPLC assay is an effective tool for surveying parasitic dynamics in an estuarine ecosystem.
Tremendous endeavors have been carried out to monitor coastal ecosystem pollution. Because the impact on humans and ecosystems is ambiguous, biomonitoring such as the ‘Mussel Watch’ Program (Kim et al., 2008), is a powerful method to measure the dynamics of lethal chemicals in the environment. Thus far, to assess the pollution level, many species have been subjected to biomonitoring programs, including micro/macrophytes (Ali et al., 1999; Burridge and Bidwell, 2002), zooplankton (Ritterhoff and Zauke, 1997; Kahle and Zauke, 2003), bivalve mollusks (Boening, 1999; O'Connor, 2002; Cho 2006), fish (Cho et al., 2003; Van der Oost et al., 2003 ). Grass shrimp
The grass shrimp is also well known as an intermediate host for parasites such as, the trematode
Recently, denaturing high-performance liquid chromatograph (DHPLC) has been introduced in marine biology as a versatile and competent tool for the identification of planktonic bivalve larvae (Wang et al., 2006) and bacterial composition in environmental samples (Barlaan et al., 2005). The DHPLC technique has also been also successfully used to detect parasites in blue crabs (Troedsson et al., 2008a, 2008b). The 18S ribosomal subunit has been used as an attractive target for DHPLC assay because it contains highly conserved regions throughout eukaryotic organisms but also has many high variation regions (Troedsson et al., 2008a). The DHPLC technique can be a prominent tool for diagnosing disease or parasitic infections, especially for unidentified parasitic species. In this study, we developed a DHPLC assay by 18S universal primers to detect parasitic infection of the grass shrimp, which is an indicator species of estuarine ecosystem health.
Using isolated 18S ribosomal DNA from the grass shrimp
>
Small subunit ribosomal DNA (SSU rDNA) and sequencing
For DNA extraction, fresh muscle of grass shrimp and its parasites were carefully dissected under a stereomicroscope and then digested with proteinase-K using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) following manufacture’s instruction. Partial 18S rDNA sequences were amplified by PCR-amplification using a universal primer set: UnivF- 15 (5′-CTGCCAGTAGTCATATGC-3′) and UnivR-1765 (5′-ACCTTGTTACGACTTTAC-3′). PCR amplification was performed using a Gene 9700 Thermo cycler (Applied Biosystems, Foster City, CA, USA) with the following parameters: 95℃ for 3 min, followed by 25 cycles at 94℃ for 30 s, 45℃ for 30 s, and 72℃ for 60 s, with an additional extension at 72℃ for 7 min.
After gel electrophoresis using a 1% agarose gel, the 18S rDNA fragments were purified using Quantum Prep Freeze ‘N Squeeze DNA Gel Extraction Spin Columns (Bio-Rad, Hercules, CA, USA) and then cloned using the TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA, USA). Plasmid DNA was extracted and purified using a QIAprep Spin Miniprep Kit (Qiagen). Sequence analysis was performed using an automated capillary sequencer (CEQ-8000; Beckman Coulter, Fullerton, CA, USA) with a CEQ DTCS Quick-Start Sequencing Kit (Beckman Coulter) and M13F and M13R sequencing primers. Sequences were assembled using ContigExpress of Vector NTI Advance 10.3.0 (Invitrogen). Phylogenic analysis was also carried out using Basic Local Alignment Search Tool (BLAST; Zhang et al., 2000), and a phylogenic tree was constructed using the pairwise alignment neighbor-joining (NJ) method.
>
PCR amplicons for the DHPLC assay
To develop the DHPLC assay, 18S rDNAs of each species were amplified using a set of universal primers: UnivF-1131 (5′-AAACTYAAAGRAATTGACGG-3′) and UnivR-1629R (5′-GACGGGCGGTGTGTRC-3′). The PCR reaction mixture contained 0.5 μM of each primer, 200 μM of dNTP (Promega, Madison, WI, USA), 0.3 unit of Discoverase dHPLC DNA polymerase (Invitrogen), 2 mM of MgSO4, and 1× Discoverase PCR buffer (Invitrogen) in a final volume of 20 μL with 2 μL of 25 ng/μL template. PCR amplification was performed using the following steps: 94℃ for 30 s, followed by 25 cycles at 94℃ for 30 s, 54℃ for 30 s, and 68℃ for 30 s, with an additional extension at 68℃ for 5 min. Following electrophoresis using a 1% agarose gel, the PCR amplicons were used as a template for the DHPLC assay.
>
Factors influencing chromatographic characteristics
To investigate chromatographic dynamics, the oven temperature- dependent thermodynamic characteristics of each double-stranded PCR product were tested in varying temperatures up to 75% of the helical fraction predicted by Navigator software (Transgenomic, Omaha, NE, USA).
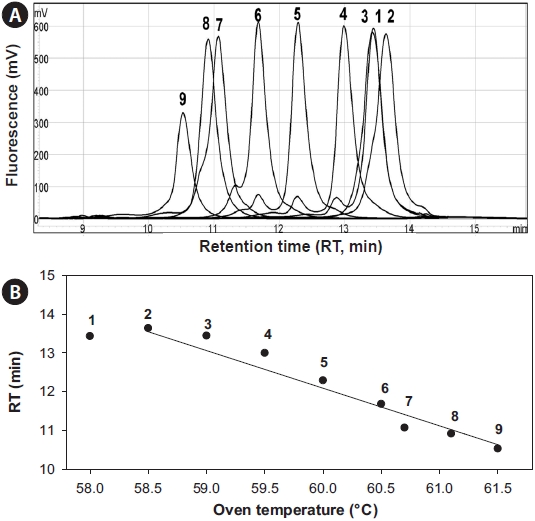
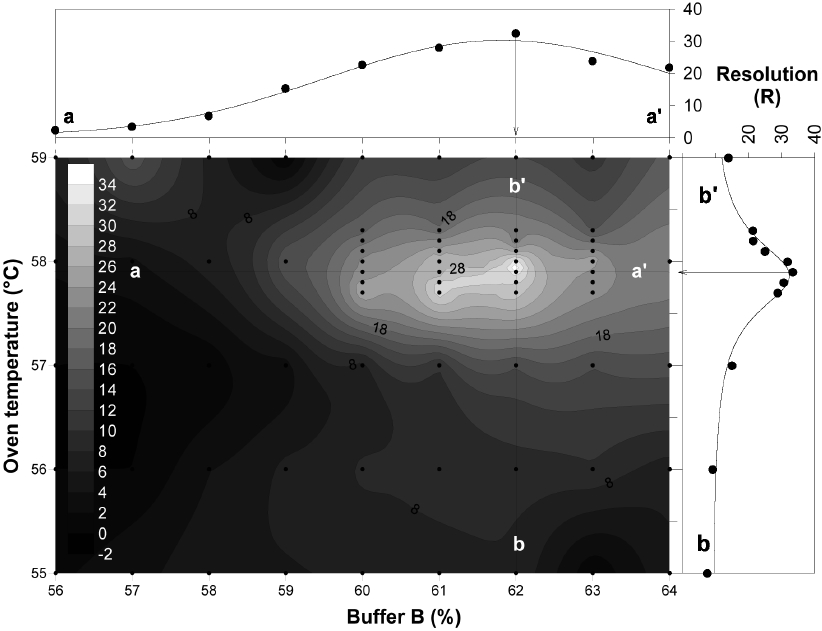
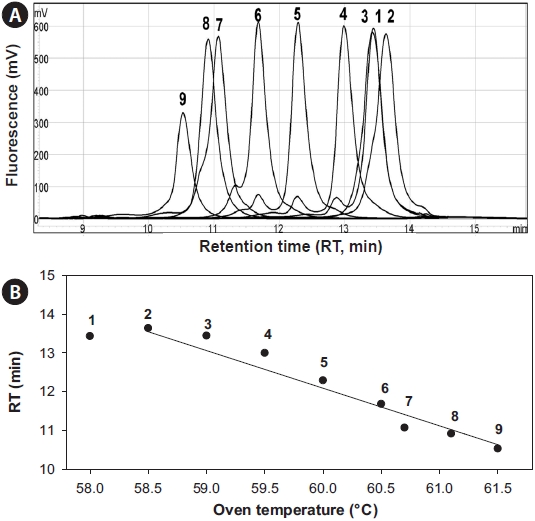
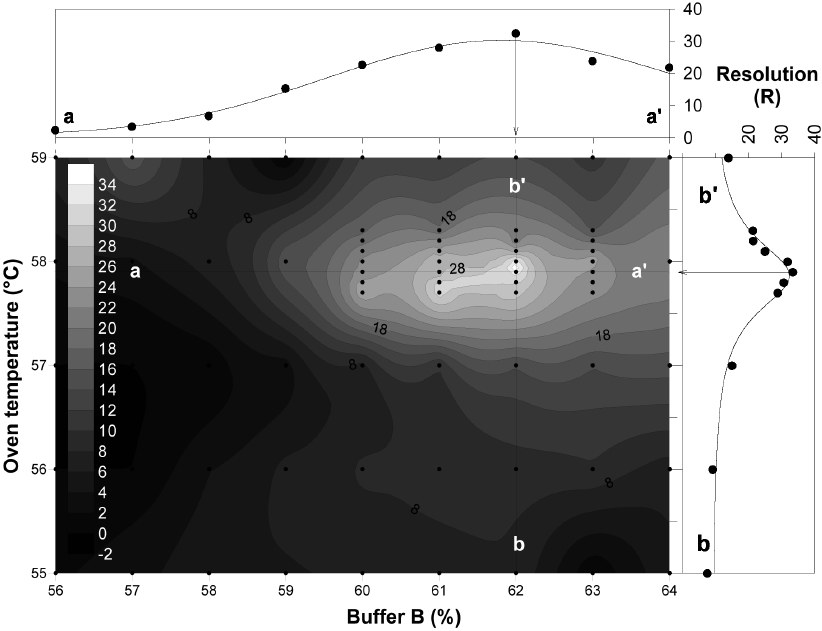
PCR amplicons were equally mixed together and then applied to DHPLC separation by varying the buffer concentration and oven temperature. Then 5 μL of template (16.5 ng of each PCR product) was injected into a DNASep HT Cartridge (Transgenomic). A systematic 11 × 9 matrix factorial analysis was conducted using 11 steps of oven temperature (55-59℃) and 9 steps of buffer B concentration (56-64%) with 68% of the fixed ending concentration. The total elution time was set as 10 min. Separation buffers were purchased from Transgenomic: WAVE Optimized Buffer A (0.1 M triethylammonimum acetate [TEAA]) and WAVE Optimized Buffer B (0.1 M TEAA in 25% (v/v) acetonitrile). The flow rate was also tested from 0.35 to 0.9 mL/min to achieve the highest resolution (
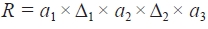
>
Development of the blocking probe
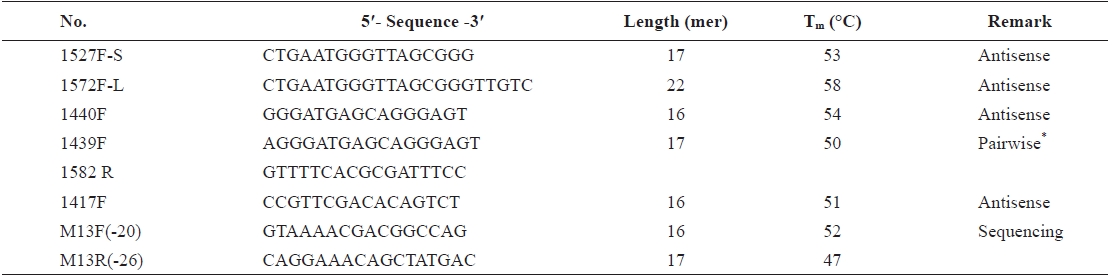
To facilitate detection of low copies of parasitic DNA sequences, species-specific blocking probes targeting 18S rDNA fragments for the DHPLC assay were designed using Primer Premier 5.0 (Biosoft International, Palo Alto, CA, USA). The blocking probe was synthesized from peptide nucleic acid (PNA), which was designed to clamp PCR primers and thus prevent the activity of polymerases. After confirmation of the sequence using C3 spacer, PNA were synthesized for the blocking probe (Panagene, Daejeon, Korea).
>
Verification of the DHPLC assay
To validate the DHPLC assay using wild samples, genomic DNA was extracted from whole bodies using liquid nitrogen grinding. Then 2 ng of extracted genomic DNA was applied to PNA-PCR in 20 μL of reaction mix containing 0.5 μM of each primer, 200 μM of dNTP (Promega), 0.3 unit of Discoverase dHPLC DNA polymerase (Invitrogen), 1× Discoverase PCR buffer (Invitrogen) containing 2 mM of MgSO4, 1 μM of the PNA probe, and 2 ng of template. PNA-PCR amplicons were separated using a DNASep HT Cartridge (Transgenomic). To identify the composition of species, the peaks eluted from DHPLC separation were collected by fraction collector (FCW- 200; Transgenomic) and applied to sequencing following the method described above. Phylogenetic analysis of each peak was performed using the BLASTN search and NJ method in GenBank (Zhang et al., 2000).
Linear regression was carried out for thermodynamic variation of chromatographs in response to oven temperatures and amounts of amplicons relative to the concentration of the blocking probe. For determining the peak of variables, a nonlinear regression for R was carried out for buffer B and oven temperature. The statistical analyses were performed using SigmaPlot (Systat Software, Inc., Chicago, IL, USA). To optimize the conditions for DHPLC separation, variables from an 11 × 9 matrix factorial experiment were analyzed against the contour of R using Surfer 8.05 (Golden Software, Inc., Golden, CO, USA).
>
SSU rDNA gene and chromatographic dynamics of single amplicons
Nearly full-length 18S rDNA sequences of grass shrimp and its parasites were PCR-amplified (1,857 bp for
To investigate the thermodynamic characteristics of the rDNA sequences, 5 μL of the 527-bp 18S rDNA amplicon of
>
Optimization of DHPLC conditions for separating the 18S rDNA amplicons
To optimize DHPLC separation, 11×9 matrix factorial analyses were carried out using two variables: oven temperature (56-59℃) and percent buffer B (56-64%). The contour analysis showed that the highest R value was achieved at about 57.9℃ and 62% of buffer B. The distribution of R was further analyzed by regression analysis to confirm the peak of R. At 57.9℃, R was nonlinearly regressed against buffer B with the highest peak at 62% of buffer (line a-a′;
>
Development of the blocking probe
To develop the blocking probe sequence, we tested several species-specific sequences of 18S rDNA of
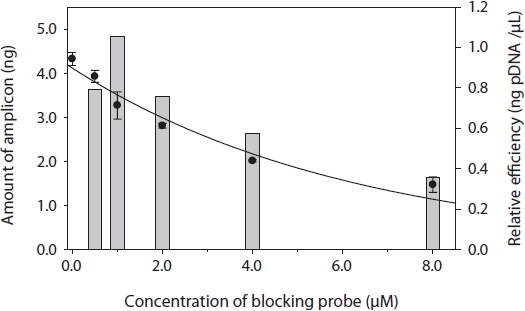
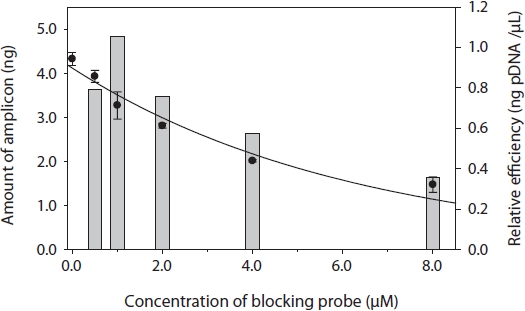
To determine the optimal concentration of the blocking probe, the blocking efficiency of PP-1417F was investigated using 0-8 μM of blocking probe and cloned plasmid DNA of
[Table 1.] Information of blocking probe candidates for PNA development
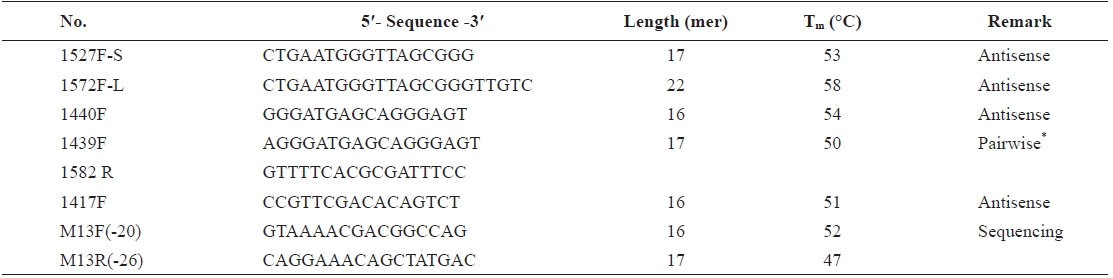
Information of blocking probe candidates for PNA development
>
DHPLC separation for the wild-type sample
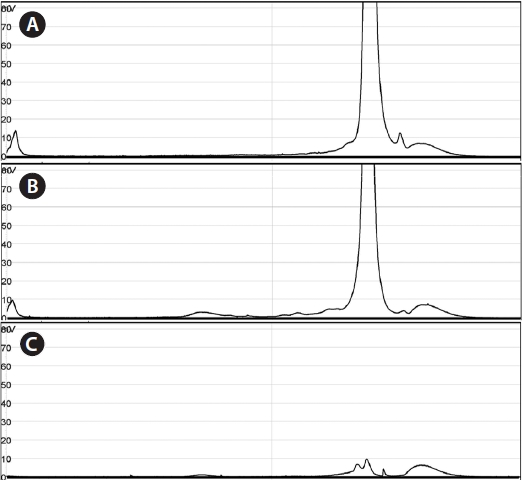
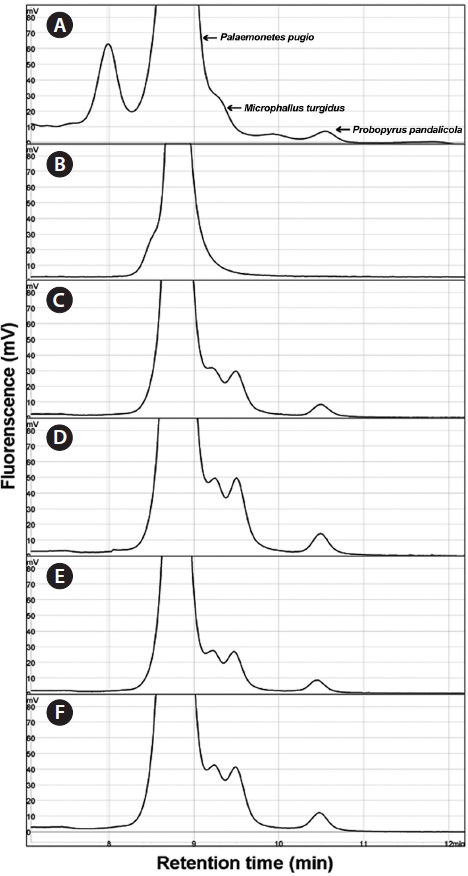
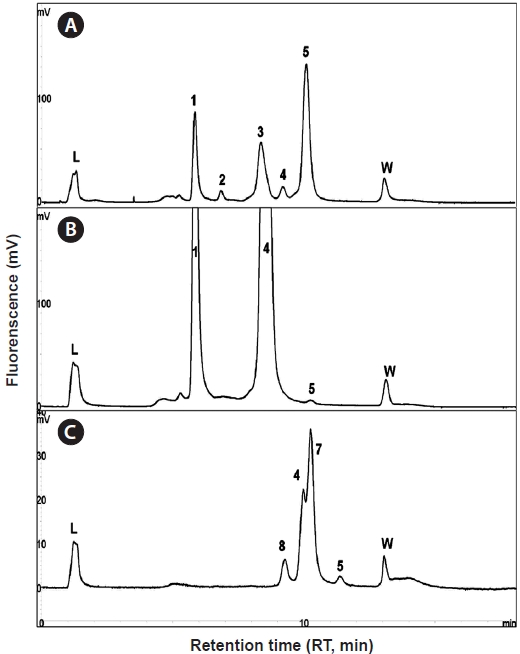
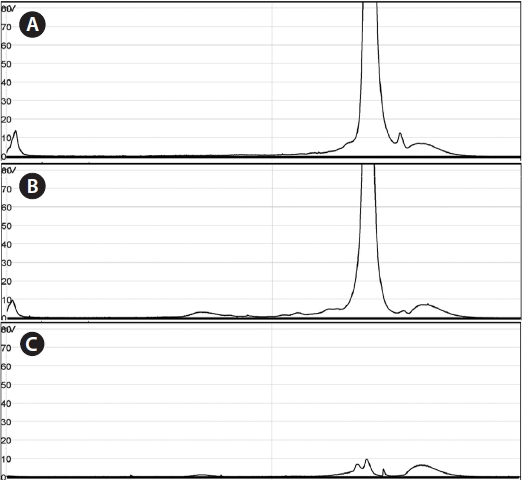
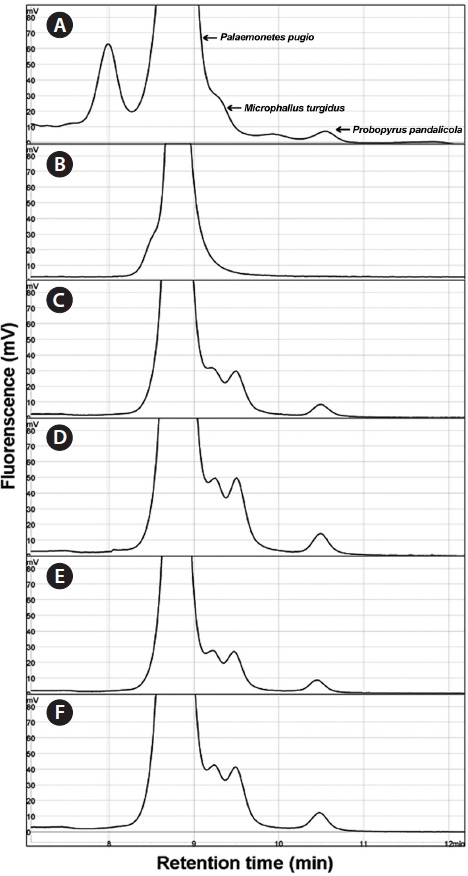
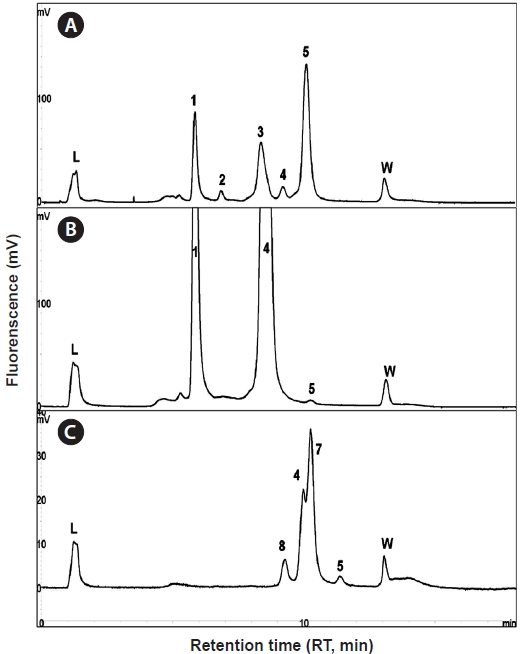
Using the optimized DHPLC assay, genomic DNA from the wild sample was applied to DHPLC separation after PNA-PCR with 1 μM of the PNA blocking probe. The PNAPCR- DHPLC assay showed enhanced PCR amplification of sequences with very low copy numbers. To evaluate our optimized DHPLC assay, each gene fragment was collected and sequenced for the species identified. PP08-116, which was infected by the bopyrid isopod
Detection of symbionts and/or parasites is crucial for understanding ecosystem homeostasis in coastal ecosystems. Recently, PCR methods have provided useful tools to address these matters and expand its application to various diseases and parasitism (Lightner and Redman, 1998). PCR diagnosis has mostly been used to evaluate the presence of DNA from
certain species with high sensitivity. To overcome these drawbacks, universal primers have been developed to target SSU rDNA of diverse phylum (Bower et al., 2004; Blankenship and Yayanos, 2005). When coupled with universal primers, DHPLC can be a competent method due to its high sensitivity for separation, which can detect as small as a 2-bp difference in sequence composition (Troedsson et al., 2008b).
Due to the ubiquitous distribution and its role in estuarine ecosystems, the grass shrimp has drawn much interest for scientists in its use as a bioindicator (Key et al., 2006). Grass shrimp is also known to serve as an intermediate host for some parasites. Therefore, the grass shrimp is a useful indicator species for monitoring parasitism in estuarine ecosystems. In this study, we developed a faster, cheaper, and simpler surveillance method compared with gel-based separation technologies used to clone plasmid DNA from model hosts and parasites.
The optimal separation conditions were determined to be an oven temperature of 57.9℃ and 62-68% of buffer B. The most important factor was the oven temperature, which skewed the regression curve more than the buffer concentration (Fig. 3), which is inconsistent with Troedsson et al. (2008a). Chromatographic behavior of each fragment was highly related with the helicity of the fragments, which suggested that <95% helicity will generate multiple peaks from a single fragment. Use of the temperature 57.9℃ accounted for 96% for
I used a PNA blocking probe to suppress PCR amplification of host genes and enhance amplification. The sequence of PNA was developed using a C3 spacer oligonucleotide because C3 is inexpensive and can be quickly synthesized. In another study, PNA showed two to three orders of magnitude greater efficiency compared with C3 in detecting sequences with very low copy numbers (Cho et al., unpublished). When developing the PNA sequence, many variables had to be considered, such as the Tm, concentration, and kinetics (Ørum, 2000). In addition, our study showed that the position of hybridization was also important to obtain a high blocking efficiency: the closer the probe was bound to the PCR primer, the greater the resulting blocking efficiency and enhanced gene amplification of very low copy number DNA fragments. Thirteen- to 18- mers of PNAs are commonly used for PNA hybridization. The concentration of PNA was tested by cloning plasmid DNA of the targeted host gene, which showed approximately 25% suppression compared to non-blocking PCR. The plasmid concentration for this experiment was approximately 106 copies, which is reasonable compared to PNA-PCR for genomic DNA. In PNA-PCR, we used 2 ng of genomic DNA as a template. In the chromatograph of PP08-116, the peak area of targeted gene fragments was much smaller than the peaks of other parasitic genes, regardless of cycle numbers, which were suppressed by 90% compared to the peak of
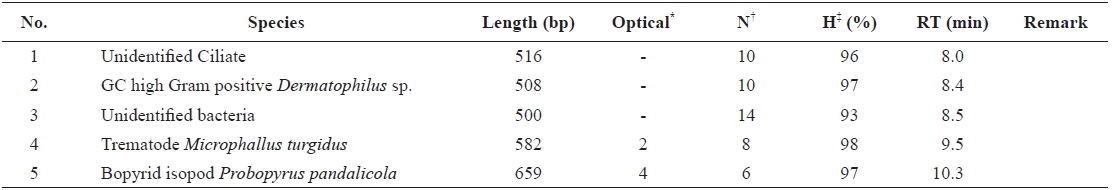
[Table 2.] Detection of symbionts using the developed DHPLC assay
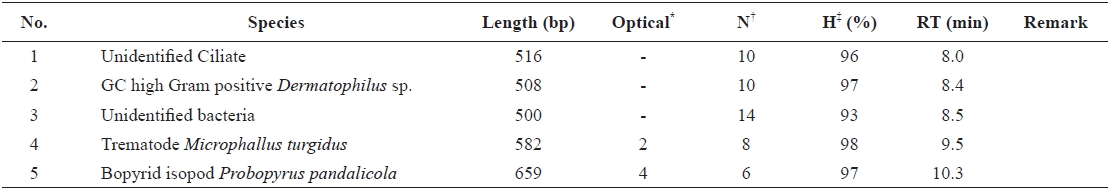
Detection of symbionts using the developed DHPLC assay
I used a universal primer set targeting highly conserved regions of 18S rDNA for PCR amplification. This universal primer set has been utilized as reliable primers from several studies (Gruebl et al., 2002; Troedsson et al., 2008a, 2008b). The detection limit of PNA-PCR in this study was relatively lower than those of species-specific PCR (103-102 copies) (data not shown), which might be acceptable for a surveillance tool of parasitism because a species-specific PCR assay does not verify the existence of various parasitism infections (Burreson, 2008). Although blocking probes can suppress the amplification of host gene fragments, this does not mean that the predominant host gene is not amplified. To overcome this problem, selective targeting of universal primers or nonphylum universal primers (Bower et al., 2004), which amplify only parasites or weakly including target species, can be considered. Several phylum-specific universal primers have been developed and extensively used not only for DHPLC assays but also for several gel electrophoresis-related methods (Iwen et al., 2002; Baker et al., 2003; Blankenship and Yayanos, 2005; Sherwood and Presting, 2007). Development of phylum- specific universal primers requires massive information on related species sequences and data processing. A combination of universal primers and blocking probe can therefore provide extensive applications of DHPLC for surveillance of parasitism.
The optimized DHPLC assay showed a well-established chromatograph of isopod-infected grass shrimp with a high peak of the parasite. The peak of the host gene was suppressed over 90% more than other parasite genes. Five peaks in total were observed from PNA-PCR from genomic DNA of wildtype samples infected by isopods, including a 508-bp 16S rDNA of GC Gram-positive bacteria at 8.407 min and a 659- bp 18S rDNA of
Estuarine systems are ecologically important environments due to their species diversity and high productivity. However, the dynamics or energetics of an ecosystem have mainly focused on macrobiota (i.e., production and/or standing stock biomass) and infectious agents, which are perceived to contribute negligible biomass to ecosystems (Poulin, 1999). Recently, however, the biomass of free-living and parasitic spe-cies was estimated to exceed that of the top predators in an estuarine ecosystem (Kuris et al., 2008). This implies a high possibility of infection of estuarine living animals from diverse parasites. Therefore, the monitoring of parasitism dynamics in estuarine/coastal ecosystems can be a crucial to maintain the homeostasis of estuarine/coastal ecosystems. From this perspective, the DHPLC assay developed in this study for grass shrimp provides useful information about parasitic dynamics in estuarine ecosystems.