The genus Chlorella is unicellular green algae with sphere or elliptical shape of very small size around 2-10 μm. Since Chlorella vulgaris Beijerinck, which was a type species of the genus Chlorella was isolated (Beijerinck, 1890), a number of Chlorella were widely studied or utilized as industrial materials (Scragg et al., 2003; Yoshida et al., 2006; Rioboo et al., 2009). Most of the coccoid green algae that are very small in size and similar in shape, classification of Chlorella has been considered as one of the most difficult studies in phylogenetic classification (Krienitz et al., 2004).
Though more than 100 species of coccoid green algae were classified as members of the genus Chlorella, most of them were transferred to other genus such as Micractinium, Didymogenes and Actinastrum through taxonomic studies based on morphological characters (Fott and Novakova, 1969; Andreyeva, 1975). Because of the lack of distinct morphological characters for species for identification of Chlorella, the ultrastructure of cell walls or pyrenoids (Ikeda and Takeda, 1995; Nemcova and Kalina, 2000), comparative physiology and biochemistry (Kessler and Huss, 1992; Kadono et al., 2004), and nutrient requirements (Shihira and Krauss, 1965; Wang and Dei, 2001), were often adopted for their precise classification.
Since the first report of 18S ribosomal RNA gene (rDNA) sequence of the type species, C. vulgaris (Huss and Sogin, 1989) the polyphyly of the genus Chlorella was proposed based on 18S rDNA sequences information (Huss and Sogin, 1990; Friedl, 1997; Huss et al., 1999). After that, through accumulated biochemical data and analysis of 18S rDNA sequences, only four “true” Chlorella species (C. kessleri, C. lobophora, C. sorokiniana, and C. vulgaris) was proposed (Huss et al., 1999). Besides, on the basis of the phylogenetic analysis of 18S rDNA sequences, Krienitz et al. (2004) had suggested that the family Chlorellaceae is composed of Chlorella and Parachlorella clades. More recently, Luo et al. (2010) revealed that five genera of green algae (Micractinium, Didymogenes, Actinastrum, Meyerella, and Hegewaldia) were phylogenetically closely affiliated to the genus Chlorella inferred from nucleotide sequences of 18S rDNA and ITS regions.
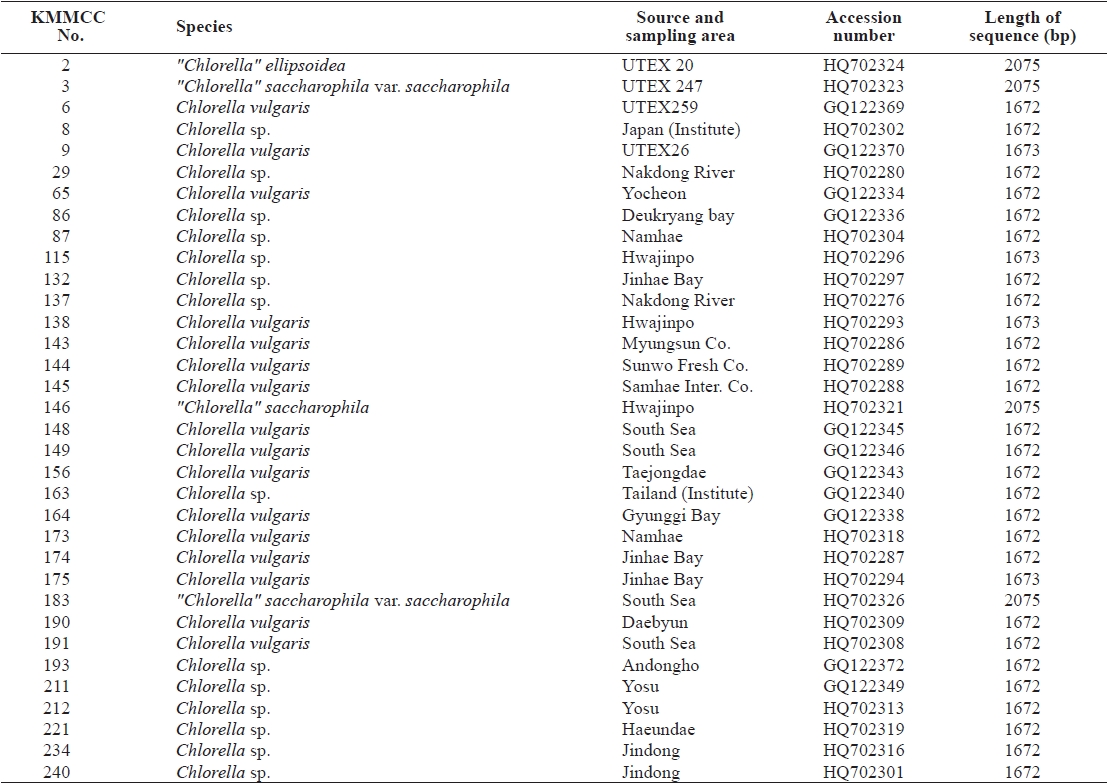
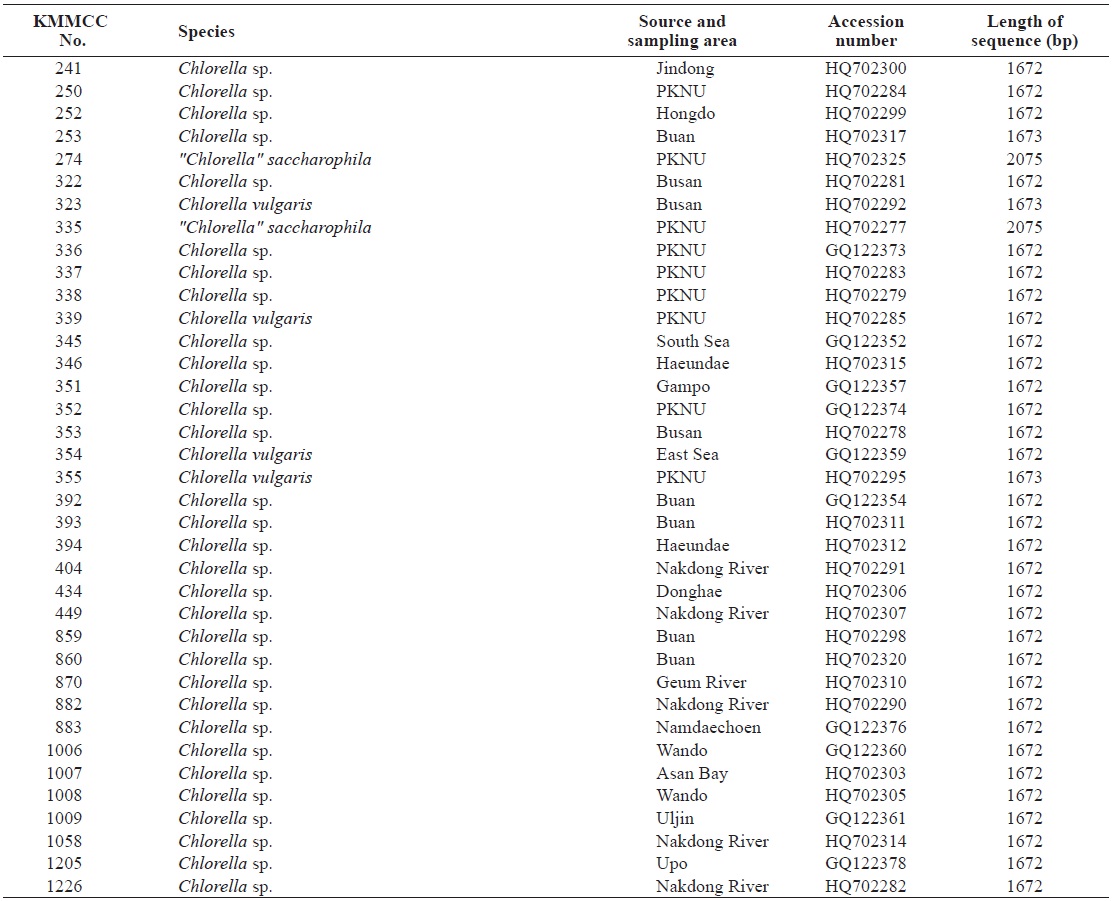
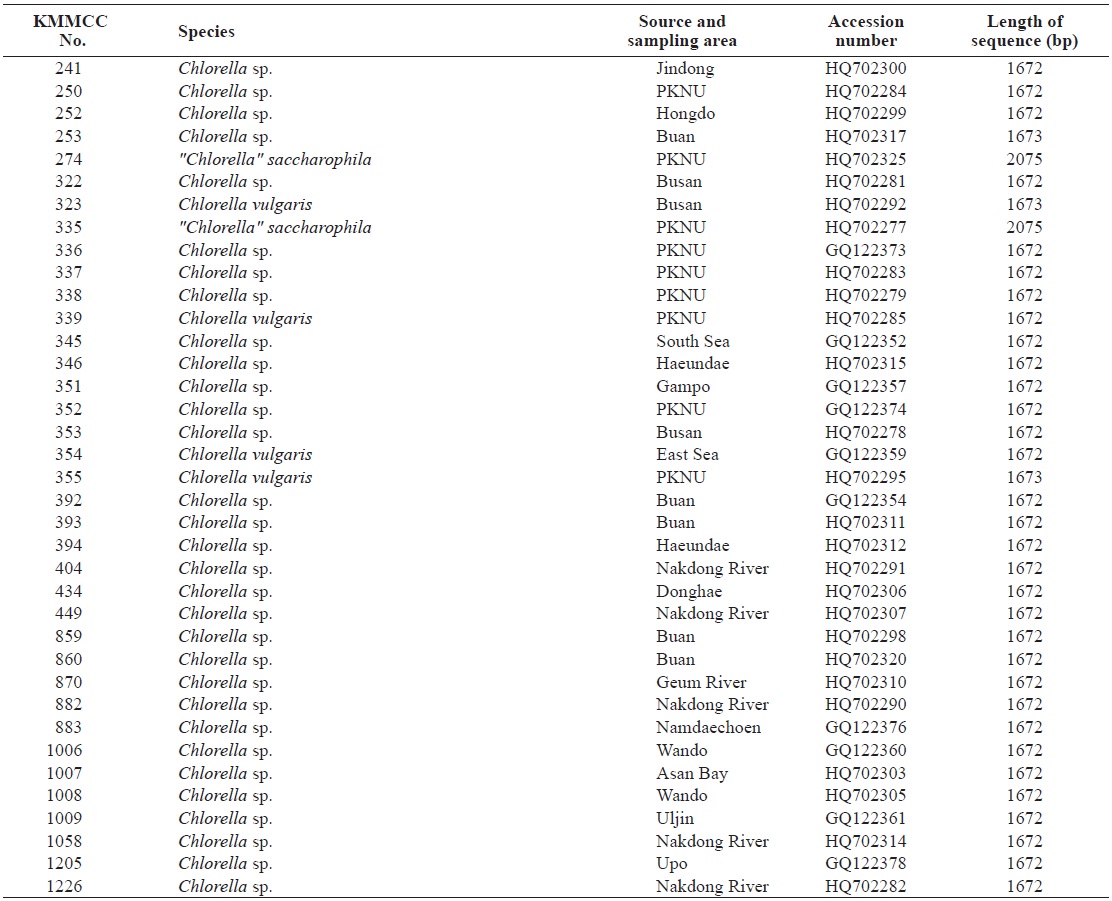
Korea Marine Microalgae Culture Center (KMMCC) had collected 80 species of Chlorella-like species from coastal and inland regions in Korea or from foreign collection centers (Hur, 2008). Chlorella-like species were identified at level of genus by observation of light microscopy, but it was confusing to identify at species level because of their similar morphology. In particular, Chlorella-like species isolated from estuary water was obscure to identify its origin as marine or freshwater
species. Therefore, in this study, 18S rDNA sequences of 71 Chlorella-like species collected mainly from Korean waters were examined. In addition, growth and size of representative Chlorella-like species from each identified group were also tested at different salinities of culture media. And the correlation between molecular phylogenetic relationships and phenotypic characteristics on growth and size of Chlorella-like species was analyzed.
Seventy-one Chlorella-like species used in this study were received from KMMCC in Pukyong National University. Among them, six species were obtained from abroad and the rest of the 65 species were isolated from coastal and inland regions in Korea (Table 1). Forty six species of green algae which were used in the phylogenetic tree which were obtained from GenBank.
Chlorella-like species were stationary cultured in 150 mL media volume of 250 mL Erlenmeyer flask using a temperature of 25℃ with continuous illumination of 100 μmol photons m-2s-1 for 2 weeks. F/2 medium (Guillard and Ryther, 1962) for marine Chlorella-like species and Jaworski medium (Thompson et al., 1988) for freshwater Chlorella-like species were used. Cultured microalgae were harvested and their genomic DNA was isolated using LiCl method (Hong et al., 1995) or Wizard® Genomic DNA Purification System (Promega, Medison, WI, USA). Isolated genomic DNA was performed polymerase chain reactions (PCR) using P2F (GGC TCA TTA AAT CAG TTA TAG) / MF (ACC TGG TTG ATC CTG CCA G) forward primers and P2R (CCT TGT TAC GA(C/T) TTC TCC TTC) / RF (GTG AAC CTG C(G/A)G AAG GAT CA) reverse primers (Huss et al., 1999; Lee and Hur, 2009) for 18S rDNA gene amplification. Sequences were obtained from cloning and the sequencing process (Lee and Hur, 2009). Sequences were subjected to homology analysis by using Blast N program and aligned by using ClustalW2 program (Thomp-
son et al., 1994). For these species, 18S rDNA sequences were acquired and their accession numbers were registered in Gen- Bank of NCBI.
Genetic distance of sequences was calculated by Kimura 2-parameter model (Kimura, 1980) and sequence identity was carried out by Bioedit Sequence Alignment Editor version 7.0.5.3. (Hall, 1999). To analyze the phylogenetic relationships of sequences, maximum likelihood (ML), maximum parsimony (MP), and neighbor joining (NJ) analysis were conducted using MEGA v.5.0 (Tamura et al., 2011). The ML analysis was constructed based on the Kimura 2-parameter with proportion of invariable sites and gamma distribution split into five categories (K2+I+G). This model was selected by test as best-fit substitution model of nucleotide sequence data. The MP analysis was obtained using the Close- Neighbor- Interchange algorithm (Nei and Kumar, 2000) with search level 3 in which the initial trees were obtained with the random addition of sequences (100 replicates). The NJ analysis model was used for maximum composite likelihood and 2000 bootstrap replications were carried out to support the reliability of the tree and the species with similar sequences were grouped together.
Growth characteristics of the species from each group were examined and compared to each other. Two representative species showing the difference on the phylogenetic relationships of sequences from each group were selected and cultured at different salinities (0, 16, and 32 psu). The specific growth rate (s.g.r./day=3.322×log (N1/N0)/t1-t0 (N1 and N0: cell number at t1 and t0 day)) (Guillard, 1973) and size of the cell were examined. For this culture, f/2 medium (32 psu) was used for marine Chlorella-like species. For estuary and freshwater Chlorella-like species, nutrient concentrations of f/2 medium were also used and the salinity for estuary (16 psu) and freshwater (0 psu) Chlorella-like species was made with distilled water and sea water. Microalgae were cultured in 250 mL Erlenmeyer flask with 100 mL medium at 25℃ with continuous light of 100 μmol photons m-2s-1 for 10 days. Culture experiments were replicated three times. The cell number of microalgae was counted daily using a hemacytometer. The size of 40 cells at the initial and final culture days was measured using MoticamPro 205A CCD scientific camera (Motic Instruments Inc., Richomond, BC, Canada).
Data were analyzed by one-way ANOVA test, and Duncan’s multiple range test (Duncan, 1955) was applied for the significance level (P<0.05). SPSS version 10.1 (SPSS Inc., Chicago, IL, USA) was used for all statistical analysis.
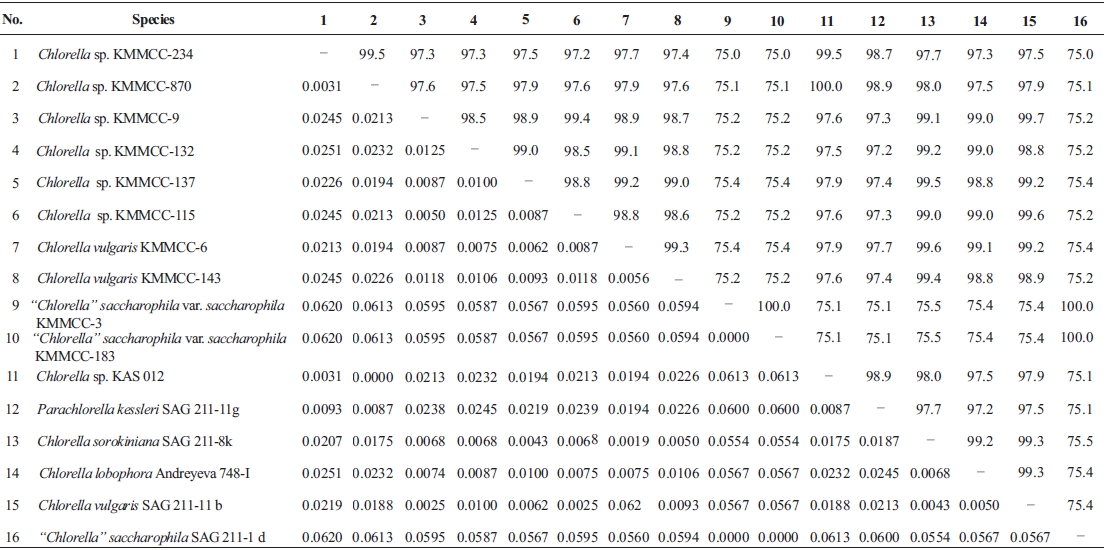
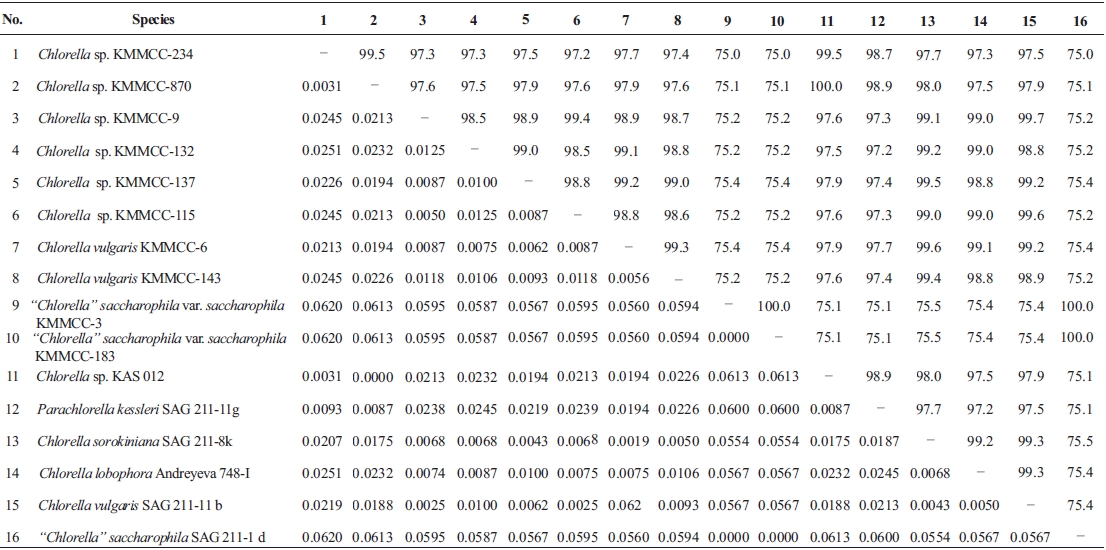
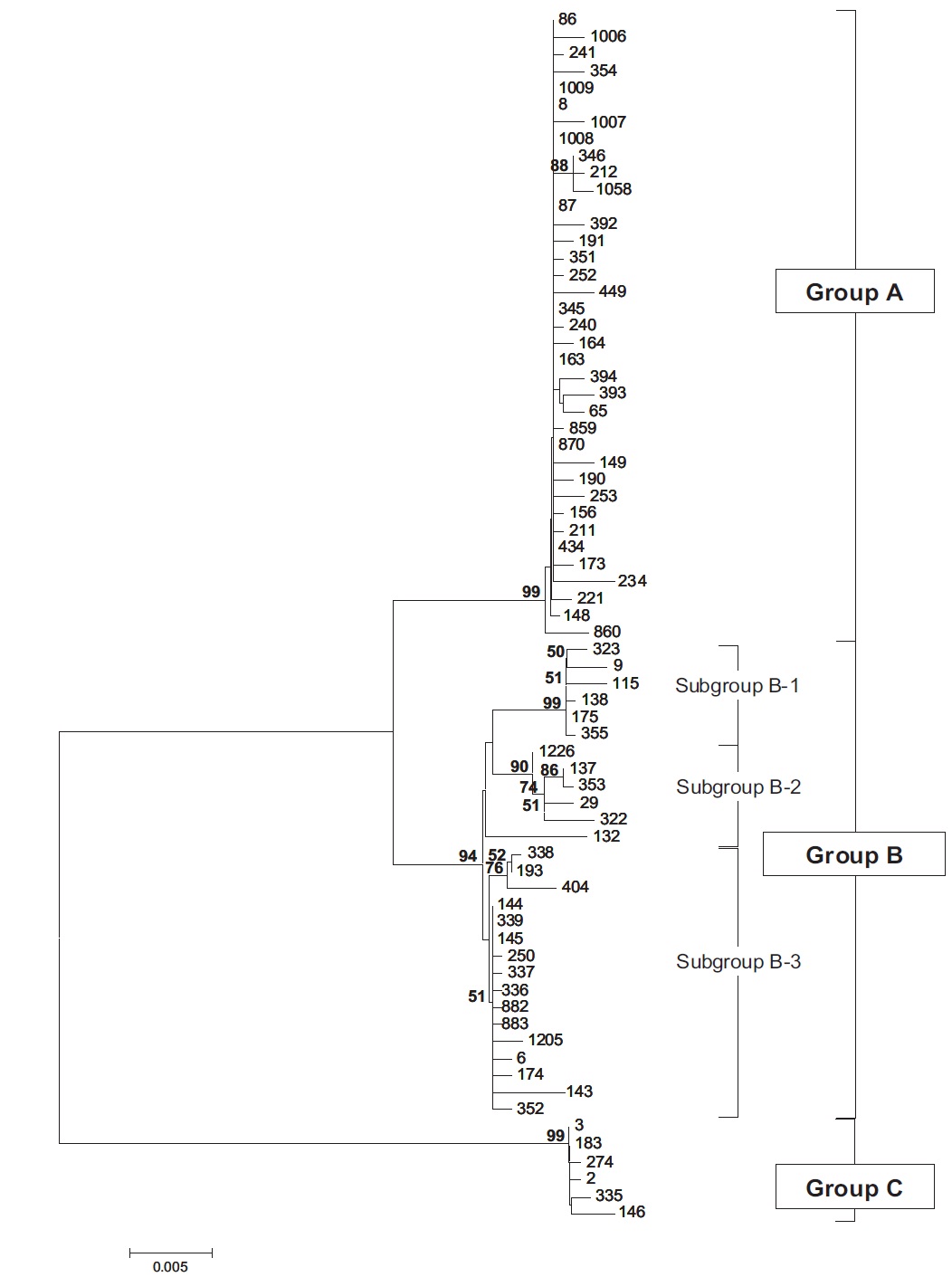
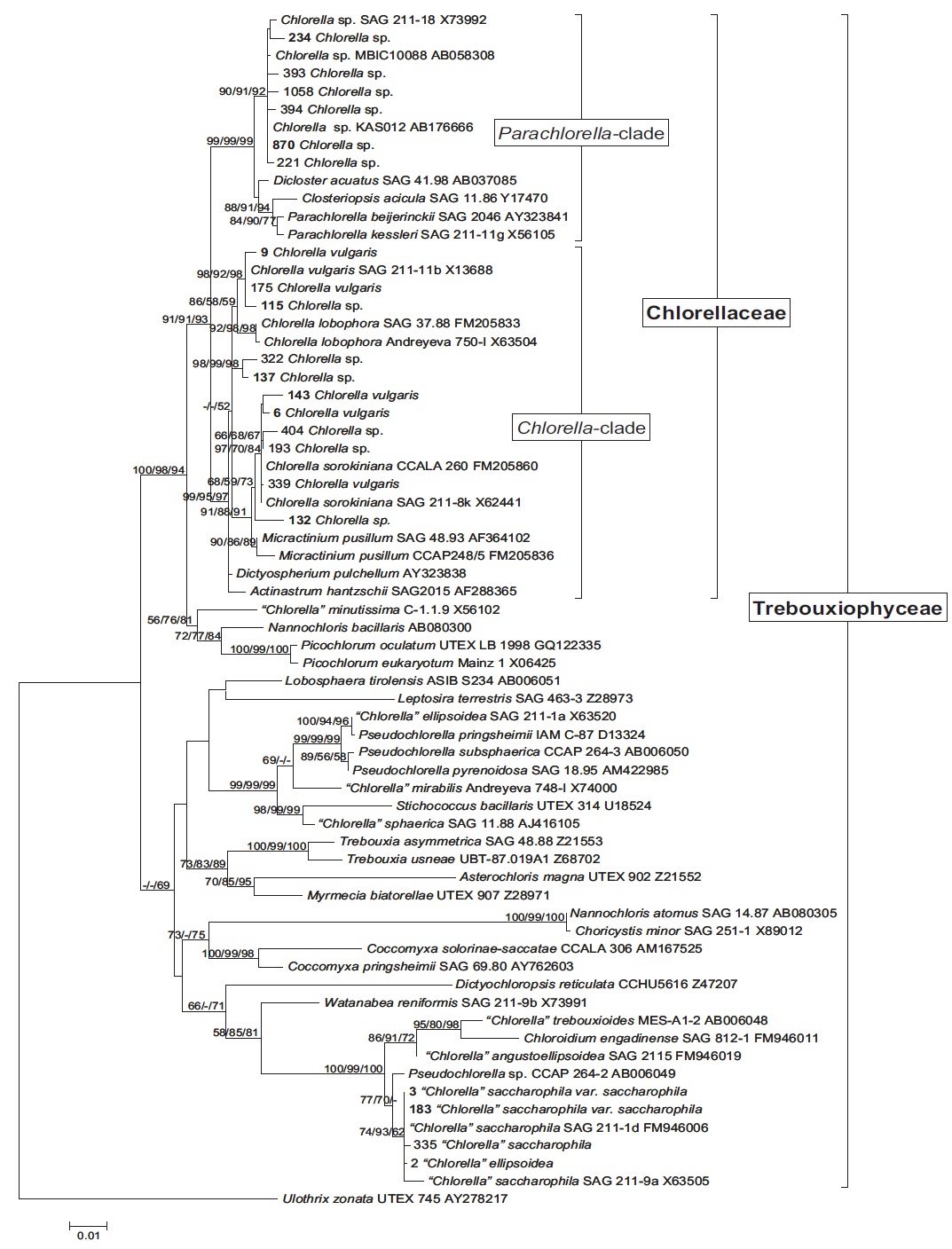
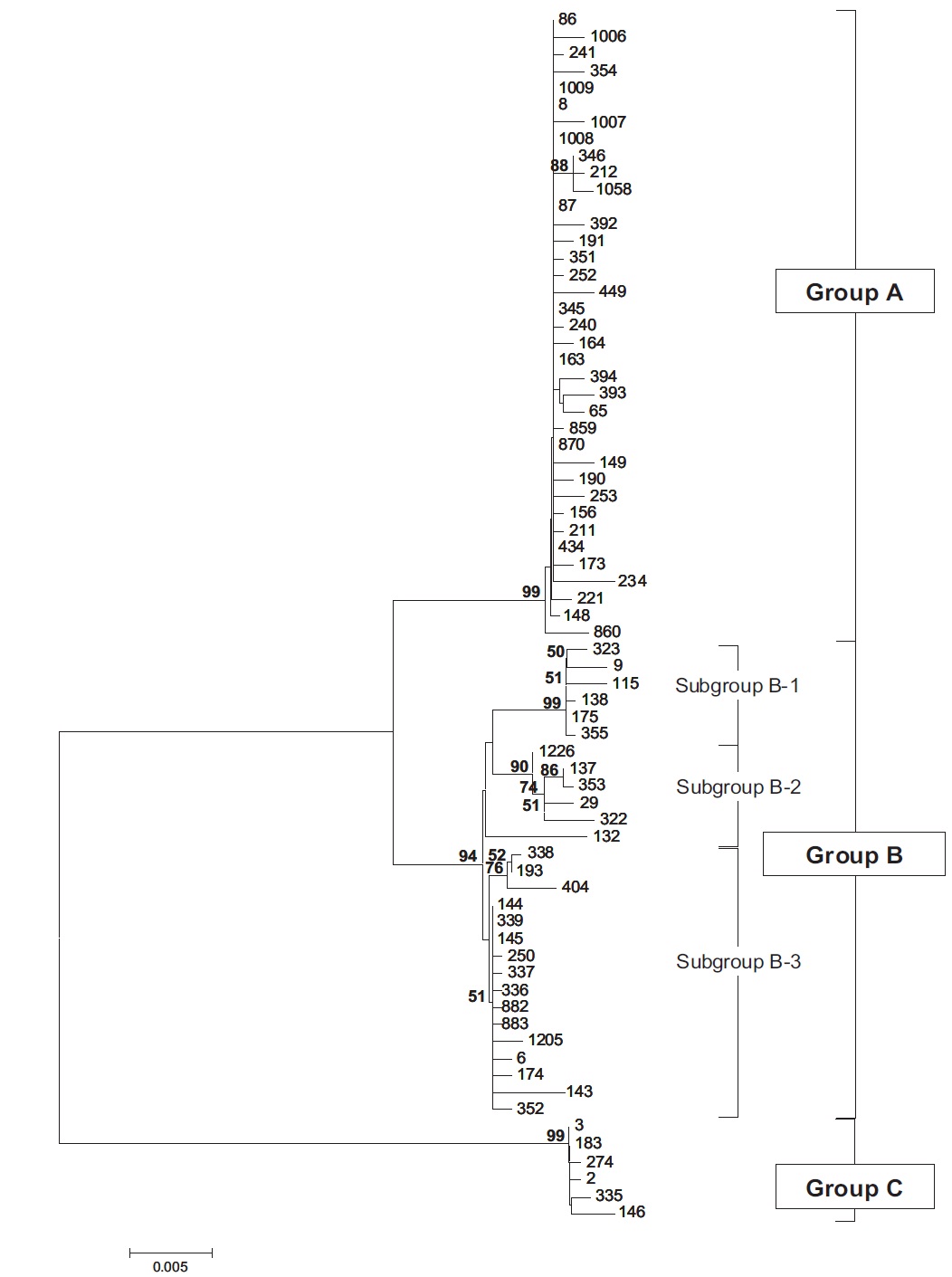
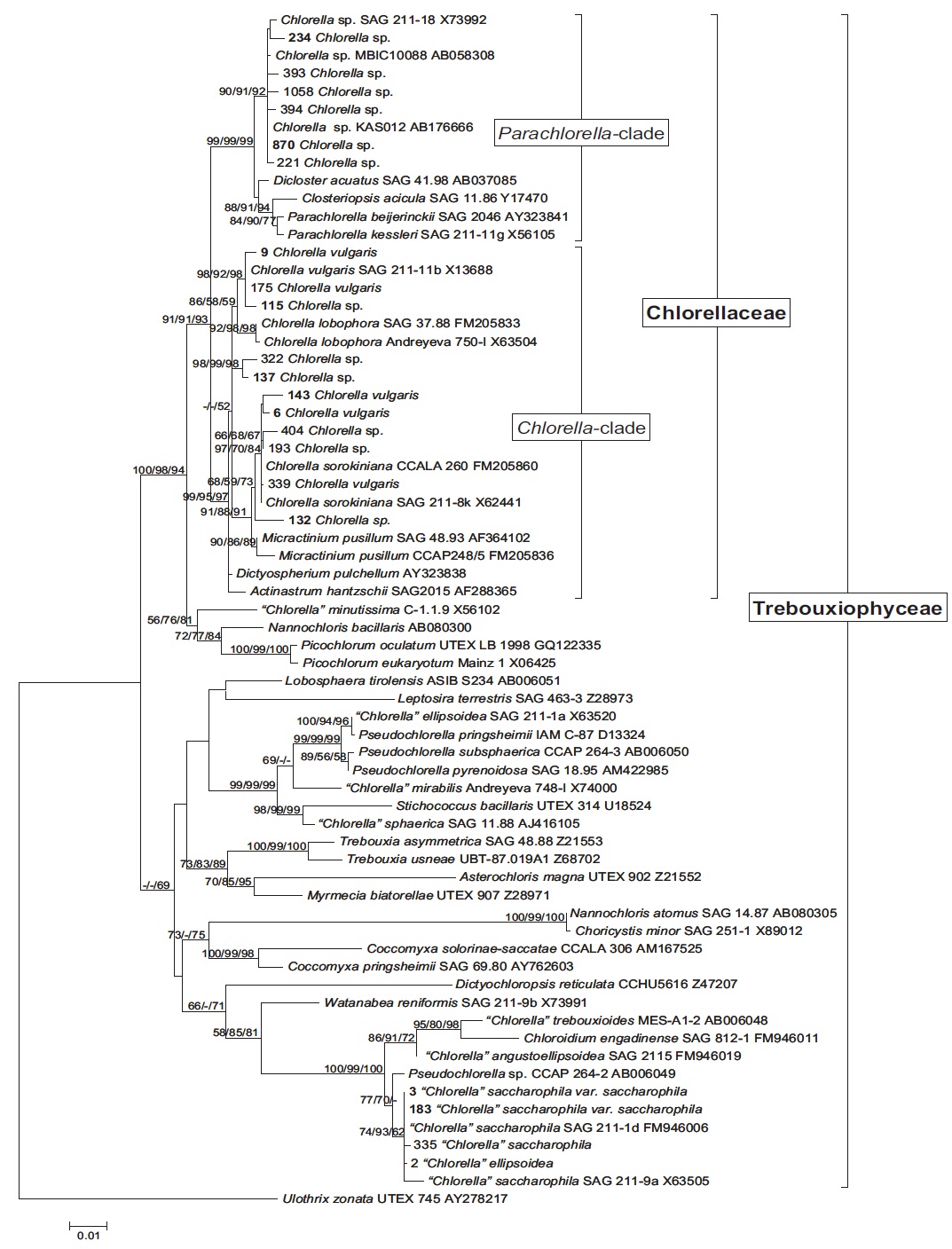
Molecular phylogenetic analysis of 18S rDNA sequences from 71 Chlorella-like species showed three independent groups (group A, B and C) with 94-99% bootstrap value. Group B was divided into three subgroups (subgroup B-1, B-2, and B-3) (Fig 1.). Maximum likelihood phylogenetic analysis by using 21 representative species of the acquired Chlorella-like species sequences and 46 species of green algae sequences referred from GenBank also confirmed three groups with 97-100% bootstrap value (Fig. 2). Sequence identity and genetic distance of 10 species (group A: KMMCC-234, 870; group B-1: KMMCC-9, 115; group B-2: KMMCC-132, 137; group B-3: KMMCC-6, 143; group C: KMMCC-3, 183) which were composed with two representative species from each of the five groups as well as six species obtained from GenBank were analyzed (Table 2). The sequence identity of KMMCC-234 and 870 from group A was 99.5%. Sequence
identity compared with group A and group B showed 97.2- 97.9%, and that between KMMCC-3 and 183 from group C turned out to be same. However, group C showed low sequence identity of 75.0-75.4% with group A and B.
Group A was diverged monophyletic with bootstrap value of 99% in Fig. 2. Thirty-seven species which corresponded to 52.1% of total were included in this group. Most of the species collected from Korean coastal waters belonged to this group. The length of 18S rDNA sequences of the species in group A was same with 1672 bp. Nine species (KMMCC-8, 86, 87, 163, 345, 434, 870, 1008, and 1009) which composed 24.3% of group A were identical in sequence and other 28 species showed sequence differences of 1-7 bp. The representative species (KMMCC-234 and 870) of group A were located in same clade with Parachlorella beijerinckii and P. kessleri and sequence identity between genus Parachlorella and species of group A was 98.7-98.9% with sequence differences of 15-21 bp. However, genus Parachlorella was closer to Closteriopsis acicula and Dicloster acuatus than group A and formed different cluster from group A. Moreover, Parachlorella was freshwater species, but most of species of group A were marine.
Chlorella sp. KAS 012 isolated from Korean sea water showed high sequence similarity over 99.5% with group A. This species exhibited very small and simple spherical morphology that was similar with the strains of Parachlorella (Aslam et al., 2007) although several egg-shaped cells of Parachlorella were observed (Krienitz et al., 2004). And Chlorella sp. KAS 012 possessed halotolerant and thermotolerant features. Aslam et al. (2007) proposed Chlorella sp. KAS 012 to genus Marinichlorella on the basis of the phenotypic and phylogenetic data. Therefore, it was judged that marine habitat species existed in Parachlorella clade. Chlorella sp. KMMCC-870 which was isolated from estuary of Geumriver was considered as a freshwater species on account of low salinity of sampling area. But this species was revised as a marine species. This misunderstanding was due to tidal currents in the estuary area.
Group B was diverged with 94-99% bootstrap value and 28 species which composed of 39.4% of total species were included in this group. Two species from UTEX (KMMCC-6 and 9) and 26 species collected from the inland area were included in this group and their sequence length was 1672- 1673 bp. Group B was divided into three subgroups. Chlorella vulgaris Beijerinck SAG 211-11b belonged to subgroup B-1. Sequence length of six species in subgroup B-1 showed same length with 1673 bp and over 99.4% identity with only 1-4 bp sequence differences (Fig. 2). Five among six species in subgroup B-2 were collected from Nakdong-river and Namcheon- cheon. The sequence length of the five species was the same with 1672 bp and sequence identity was 99.5-99.9% with only 1-5 bp sequence differences. However, in case of Chlorella sp. KMMCC-132 collected from Jinhae Bay, sequence difference was 13 bp which had large difference. And sequence identity was also slightly low (98.8-99.2%). In comparison with subgroup B-1, it showed similar tendency with 98.5-98.9% of sequence similarity and 18 bp of sequence differences. Therefore, the position of KMMCC-132 in group B was not clear. The sequence length of 16 species in subgroup B-3 which were collected from freshwater was 1672 bp. The sequence identity was over 99.1% and sequence difference was 1-7 bp in subgroup B-3. In the molecular phylogentic tree with 71 Chlorella-like species (Fig. 1), subgroup B-1 and B-2 were closely located, but B-3 was distantly diverged. It indicated that subgroup B-1 and B-2 were closer to each other. However, in Fig. 2, subgroup B-1, B-2, and B-3 were located next to each other, together with Actinastrum hantzschii and Dictyospherium pulchellum.
Group C was branched with 99-100% bootstrap value and six species of “Chlorella” saccharophila were included in group C. The sequence length of them was 2075 bp and sequence identity was 99.6-100% with 1-5 bp of sequence differences. Group C included group I intron of 400 bp sequences and neighbored with Watanabea reniformis (Hanagata et al., 1998) forming a monophyletic lineage within Trebouxiophyceae. Group C was also considerably apart from group A and B in the tree. Friedl (1995) established new class Trebouxiophyceae by analysis of the ribosomal RNA sequences from coccoid green algae, which was the sister group of Chlorophyceae. Huss et al. (1999) reported four species of ‘true’ Chlorella belong to familiy Chlorellaceae in class Trebouxiophyceae. “Chlorella” saccharophila did not belong to the true Chlorella group (Huss et al., 2002).
In this study, group A and B belonged to Chlorellaceae which were diverged with 91-93% high bootstrap value in the phylogenetic tree (Fig. 2), but the another group of Trebouxiophyceae was diverged with 49-69% low bootstrap value. This finding was similar to the result (45-59%) of Huss et al. (1999). This study also supported that “Chlorella” saccharophila of group C was not in ‘true’ Chlorella group. And taxonomic status of the group C species was unclear.
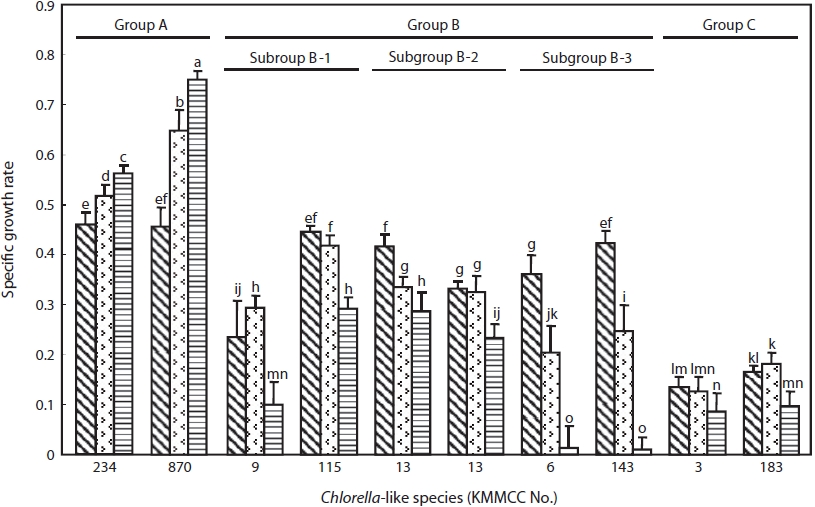
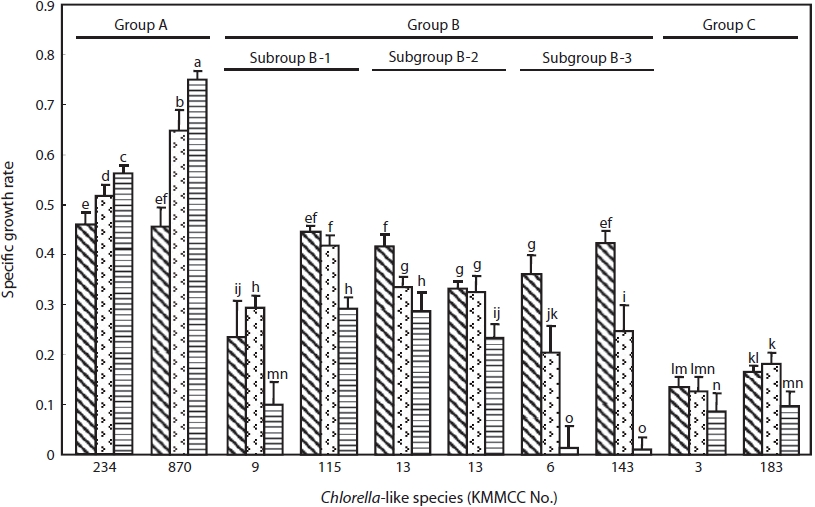
The specific growth rate by salinity of two representative Chlorella-like species from each group was shown in Fig. 3. Group A showed the highest growth rate, whereas group C showed the lowest growth rate (P<0.05). Group B showed intermediate tendency in growth rate. The specific growth rate of Chlorella sp. KMMCC-870 and 234 in group A was 0.75 and 0.55, respectively at 32 psu, which were significantly higher than that at 16 and 0 psu. The growth rate of both species at 0 psu was lowest with 0.46 (P<0.05). Six species in group B showed the fastest growth rate at 0 psu with 0.24- 0.45 compared with 0.01-0.29 at 32 psu. In particular, subgroup B-3 showed the lowest growth rate with 0.01 at 32 psu as compared with that of B-1 and B-2. KMMCC-3 and 183 in group C showed growth rate of 0.13-0.18 at 0 and 16 psu, which were the lowest among the ten species. These species
showed significant high growth rate at 0, 16 psu rather than 32 psu (P<0.05).
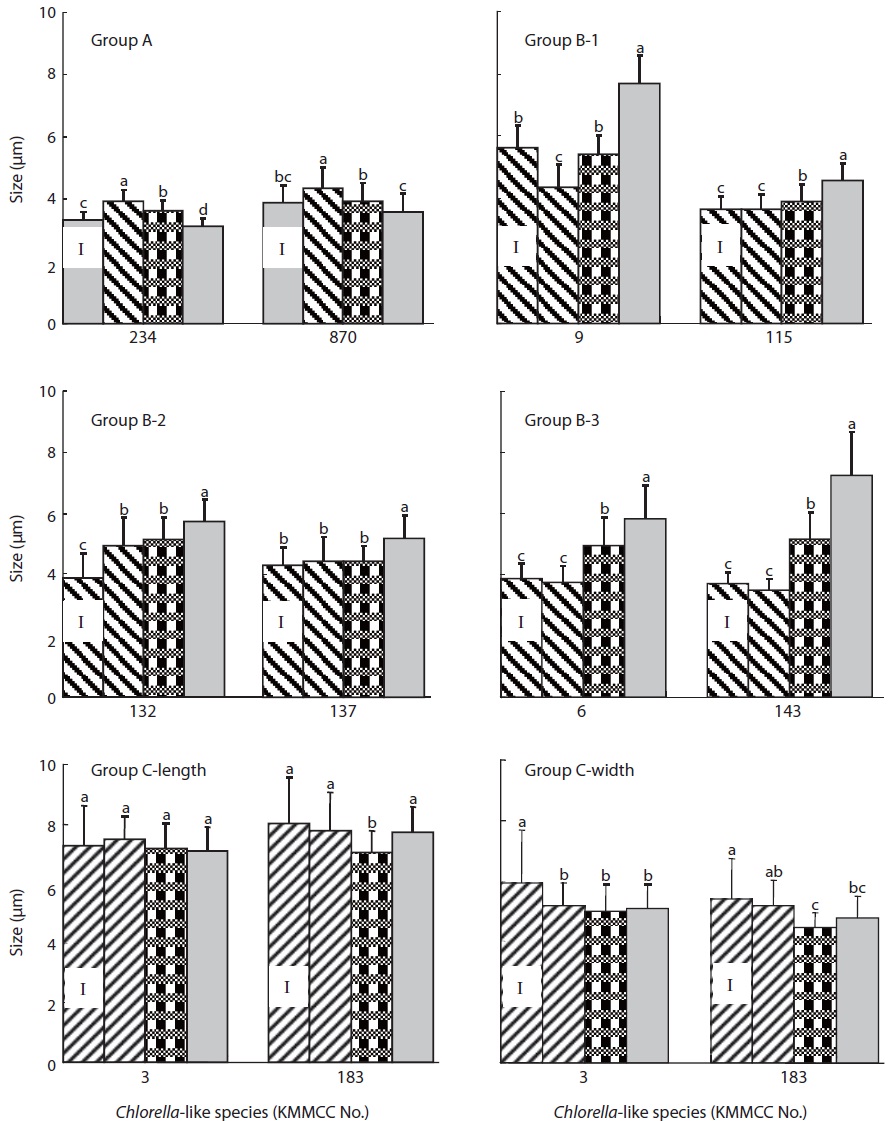
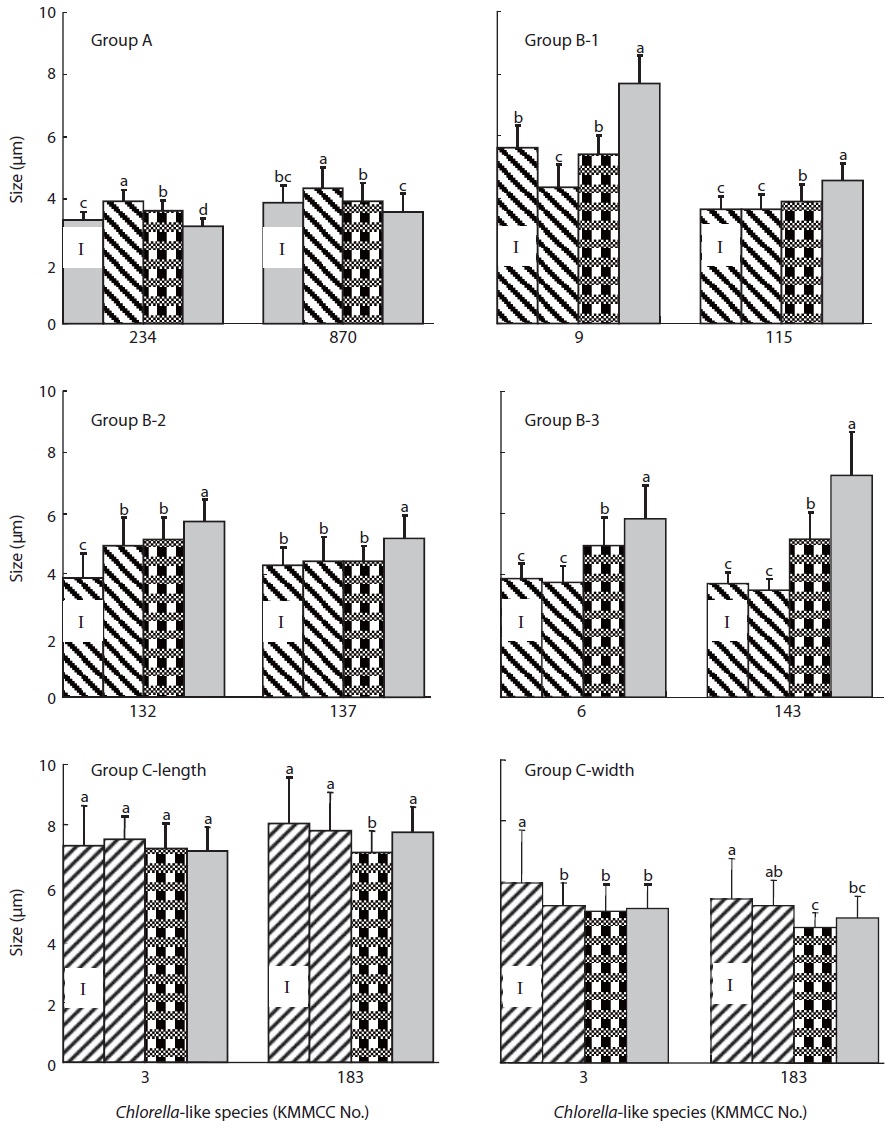
With regards to cell size after 10 days of culture at different salinities (Fig. 4), the size of KMMCC-234 and 870 in group A were 3.12 μm and 3.60 μm, respectively at 32 psu. However, the cell sizes of two species at 16 psu were 3.63 μm and 3.94 μm, and those were 3.94 μm and 4.36 μm at 0 psu, respectively. The cell size of KMMCC-234 and 870 was significantly larger in lower salinity (P<0.05). The size at the final day showed a decreased tendency compared to that at inoculation day at 32 psu. In the case of KMMCC-870 which was collected at the Geum-river, the cell size was larger as with its lower salinity. This result was also in conformity with molecular phylogenetic analysis and cell growth rate as salinity, suggesting that it was originally a marine species.
The cell size of six species (KMMCC-6, 9, 115, 132, 137, and 143) in group B varied from 3.49 μm to 4.93 μm at 0 psu after 10 days of culture. The cell size was 3.91-5.43 μm at 16 psu and 4.59-7.70 μm at 32 psu. Especially the cell size of C. vulgaris KMMCC-6 and 143 in subgroup B-3 was 3.77 μm and 3.49 μm, respectively at 0 psu. The final size of the species was not significantly different compared with initial size. But the cell sizes of them were 4.96 μm and 5.18 μm, respectively at 16 psu, and 5.85 μm and 7.25 μm, respectively at 32 psu. The cell sizes were larger at higher salinity (P<0.05). Among these, the difference in cell size of KMMCC-143 between 0 psu and 32 psu was more than two times. The cell size of the species in group A and B showed significantly larger as the conditions of salinity when compared to the optimum salinity for growth rate.
Group C showed no significant difference in cell size by sa-linity, which was contrary to that of both group A and B. This feature also corresponded with molecular phylogenetic analysis. The cell size of microalgae increased under unsuitable salinity conditions due to effect of proline (Szekely, 2004). Proline increased the size of the cytoplasm and free water contents along with glycine betain (Record et al., 1998) and engaged in increased salt resistance of microalgae (Hiremath and Matad, 2010).
Elliptical “Chlorella” saccharophila were characterized by unequal size of autospores during sporogenesis (Fott and Novakova, 1969). This feature has the advantage of survival under unsuitable environments (Darienko et al., 2010). Bigger autospores increased proliferation rate than smaller ones, while smaller ones were widely spread by the wind. So, “Chlorella” saccharophila were found in a wide range of extreme environmental conditions such as exposure to trees, rocks, and acidic soil or high temperature soil (Kessler, 1965; Huss et al., 2002; Mikhailyuk et al., 2003; Gray et al., 2007). Because of the morphological and taxonomical differentiation from ‘true’ Chlorella, Darienko et al. (2010) proposed that “Chlorella” saccharophila be transferred to the Chloroidium saccharophilum.
In conclusion, the three groups of Chlorella-like species based on the sequence analysis of 18S rDNA showed corresponding tendency with growth and size variation of the cell cultured at different salinities. Thus, the comparison between phylogenetic relationships based on 18S rDNA sequences and phenotypic growth characteristics on salinity in the culture experiment can be considered as an useful method for systematic classification of Chlorella-like species which are difficult to identify.