Use of microfluidic cell culture systems has been widespread for the investigation of precise
One of the issues in 3D cell-based assays is the require-ment of a noninvasive analytical method that allows measurement of precise dynamics or behaviors of 3D cell cultures. Among various analytical methods, optical approaches using fluorescence indicators have consistently produced a convenient yet reliable tool for various cell-based assays[8-10]. In particular, we have developed optical analytical tools for monitoring cell status
Here, we address this limit by using a DMD-based ISFODS(DMD-ISFODS) for studying 3D cell cultures. The DMD-ISFODS is designed to provide fluorescence signals from 3D cell cultures with enhanced depth resolution by employing a DMD for fast lateral scanning. Also, the DMD-ISFODS allows us to monitor biological responses with flexible optical sectioning strength because an aperture can be adjusted during observation. For example, a DMD can be used to form multiple pinholes, multiple line slits, or an S-matrix based pseudorandom sequence to reduce photobleaching and photodamage while improving resolution via structured illumination [14-17]. A DMD consists of multiple micromirror arrays which can be controlled electronically [18, 19].Thus, lateral scanning can be implemented in a short time compared to using a motorized stage and structured wavefront modulation can be performed for flexible fluorescence microscopy. Furthermore, a DMD is compact and portable in size, making a DMD-ISFODS much smaller than one based on motorized stages. To date, DMDs have been evaluated in confocal microscopy for imaging 2D cultures [20, 21]and used in optical computed tomography of tissue samples with moderate thickness up to 20 ㎛ [22]. Also, compressive sensing microscopy based on a DMD was investigated for optical sectioning of pollen grains [23]. Time-resolved 3D confocal fluorescence microscopy was developed using a DMD with limited maximum scan thickness [24].
In this feasibility study, we have constructed a DMD-ISFODS to be installed in a conventional cell culture incubator and tested initially with a USAF target for resolution evaluation and fluorescence microbeads as cell substitutes. We have then applied the DMD-ISFODS to real-time monitoring of cell statics in 3D cell cultures.
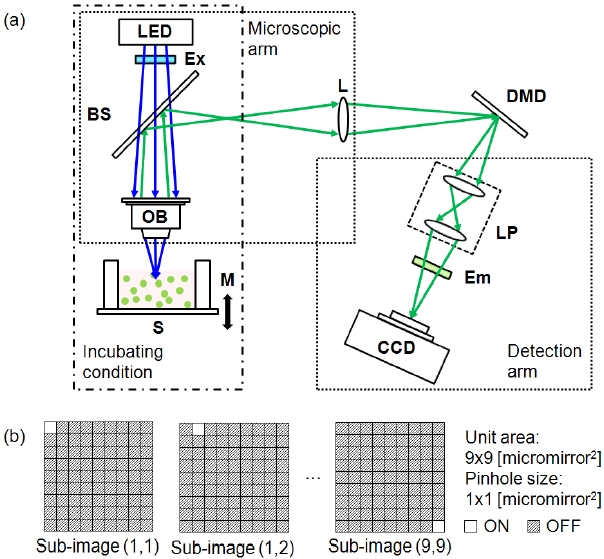
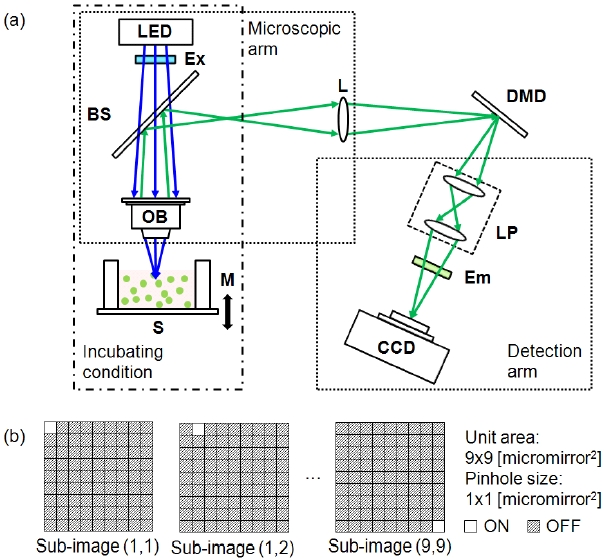
Figure 1 illustrates a simple schematic of the DMD-ISFODS.
The optical detection system consists of three parts: microscope arm, detection arm, and DMD. The microscope arm is made up of a light emitted diode (LED; λ0 = 447.5 nm, OT16-5100-RB, Rhopoint Components, Hurst Green, UK), an excitation filter (Ex; 455/70, Chroma Technology, Bellows Falls, VT, USA), a beam splitter (BS; CM1-BS013, Thorlabs, Newton, NJ, USA), and an objective lens (OB; Olympus, Tokyo, Japan). Various objective lenses were tested for optimal performance. Excitation and emission wavelengths of the DMD-ISFODS can be easily changed by replacing LEDs and filter sets. In order to extend an image focal plane to DMD, an achromatic lens (L;
2.2. Scanning and Imaging Reconstruction
Multiple pinhole apertures were used for a DMD to produce fluorescence images with fast image acquisition.In particular, 48 × 35 pinhole arrays were employed for lateral scanning. Each pinhole aperture was a single on-state DMD micro-mirror pixel sized 13.68 × 13.68 ㎛2 and scanned in a 9 × 9 matrix of a 187.14 × 187.14 ㎛2 area. The separation between pinholes is an indicator of image clarity, because pinholes farther apart would enhance depth resolution at the expense of slower scanning. For our study, pinholes were separated by 8 pixels, which was the closest distance with no visible image crosstalk. A final image was reconstructed from the intensity measured by the CCD through pinhole aperture arrays and after background correction in a 696 × 520 elements for each lateral scan. We tested two image reconstruction algorithms, (max - min) and [{summation of (image-min)} + min],
for 3D optical sectioning images [25]. After adjusting the fluorescent bead.algorithm to make it less prone to background noise, we have mainly employed the latter, which is given by
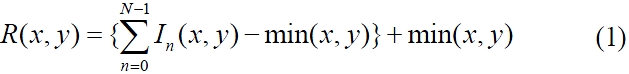
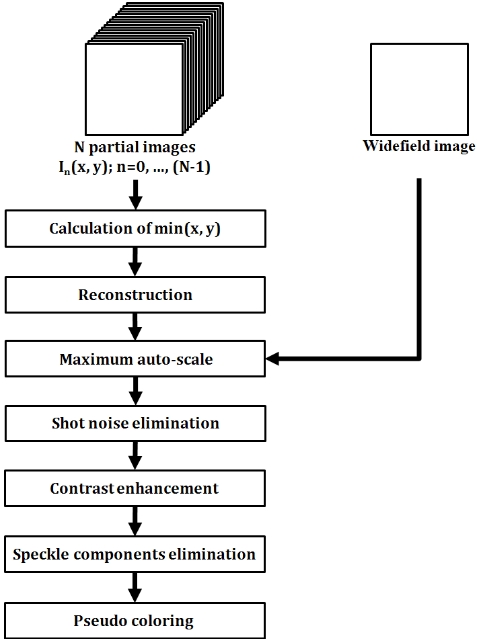
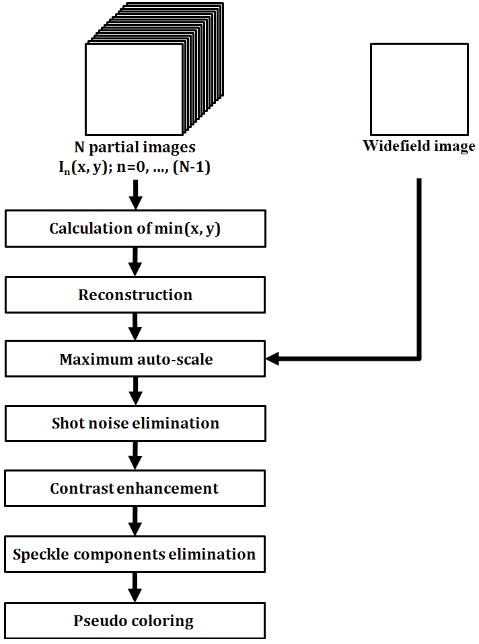
The overall image reconstruction scheme is presented in Fig. 2. Based on this scheme, the reconstruction of a single lateral image took approximately 3-4 seconds.
For the determination of the axial point spread function(PSF) of the DMD-ISFODS, measured intensity of fluorescence microbeads was deconvoluted as follows: first, we assumed the shape of a single fluorescence microbead as a perfect sphere and the emission intensity distribution as a constant.Fluorescence intensity of microbeads (diameter Φ = 10 ㎛)is then given by by
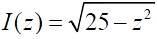
with |z| < 5. Here, z denotes the depth axis.
The 3D matrix for evaluating DMD-ISFODS was alginate-based and embedded with fluorescent beads or stained cells and was 1 mm thick in a single circular chamber fabricated on a PDMS (Polydimethylsiloxan) substrate. Fluorescent beads were sized to be 10 ㎛ in diameter (F8836, Invitrogen, Carlsbad, CA, USA). For cell imaging, we have used A431 human epidermoid carcinoma cell-line that was obtained from the ATCC (Manassas, VA, USA). Cells were cultured in T-flasks in Eagle’s Minimum Essential Medium with 10% fetal bovine serum (FBS) in a mammalian cell culture incubator
with 37℃ temperature and 5% CO2. The details of cell culture protocol and preparation of 3D cell cultures in extracellular matrix and cell culture systems are described elsewhere [13].
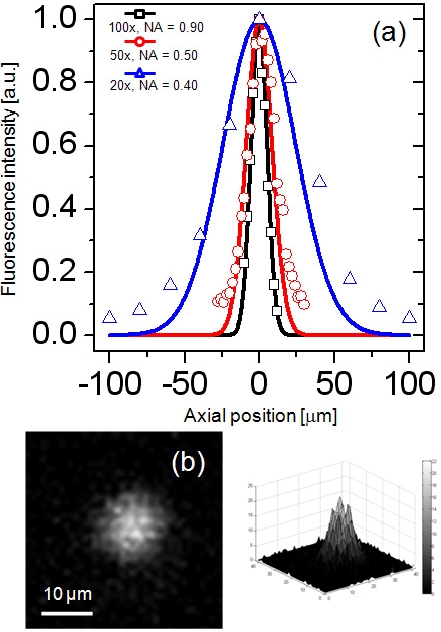
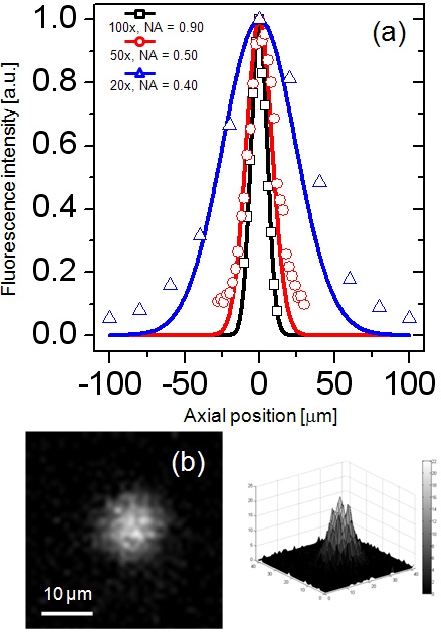
Axial PSF of the DMD-ISFODS was measured using various types of objective lenses on fluorescence beads as shown in Fig. 3. The depth of focus (DOF) was ± 6.3 ㎛ with an objective lens at 100x and NA = 0.90, which is in good agreement with the Berek formula. Overall imaging performance of the DMD-ISFODS is diffraction-limited. Note that in analyzing 3D cultures, an extremely long working distance is required because the cultures can be easily several hundred ㎛ thick while sub-cellular optical sectioning may not be critical. For this reason, we used an objective lens with lower magnification (20x, NA = 0.40) in actual acquisition of images, in which case measured DOF was ± 28.7 ㎛, because its large working distance allows depth scanning through a thick 3D culture matrix.
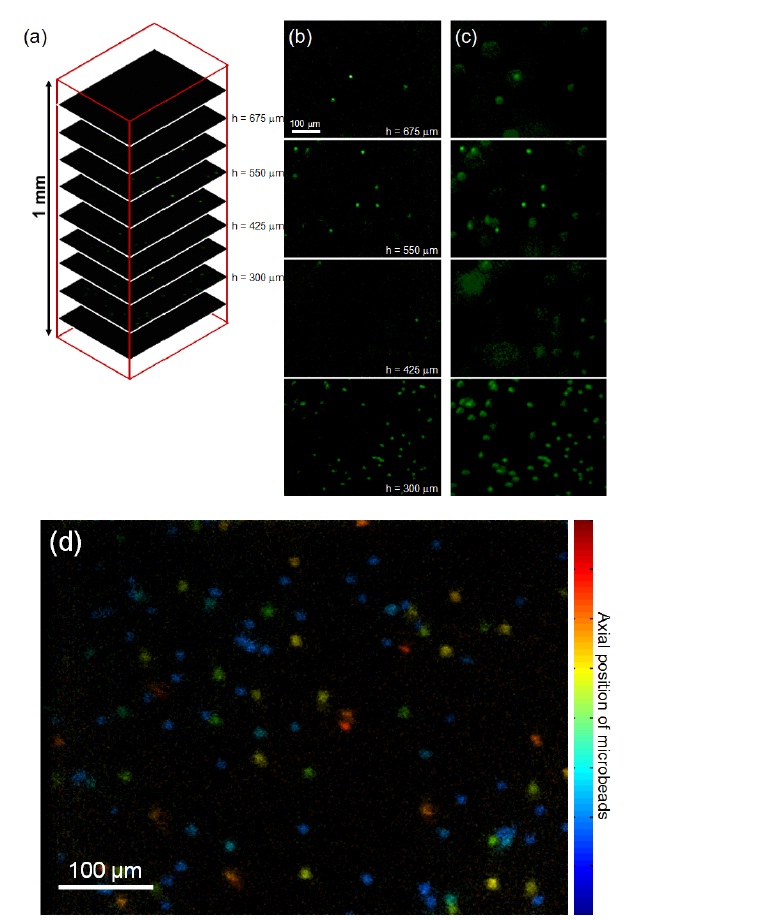
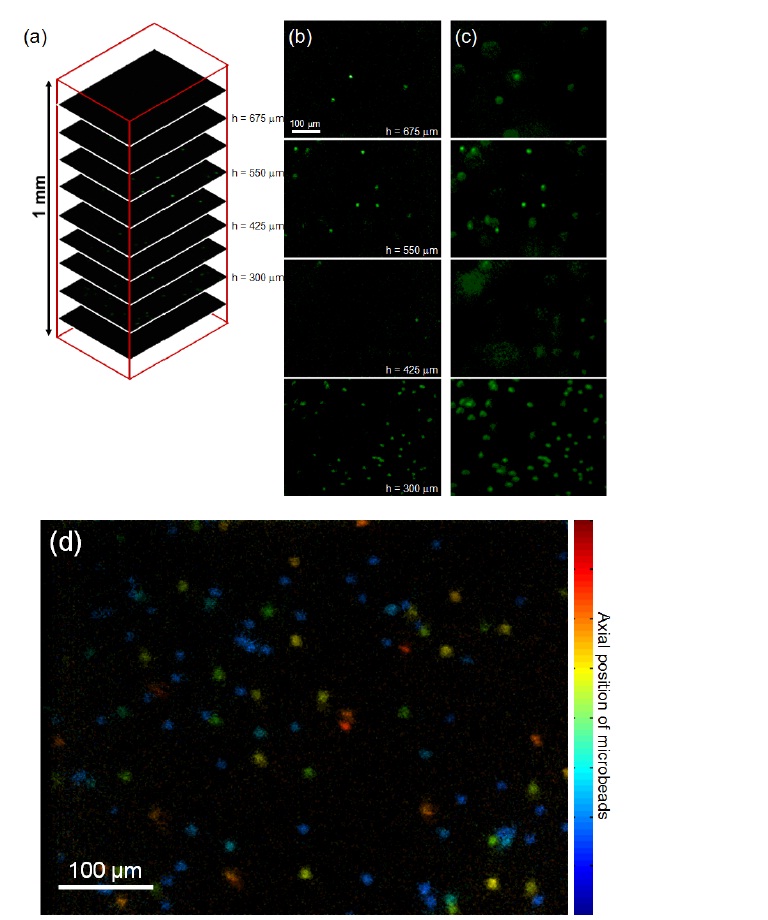
Figure 4 (a) presents the image stacks captured by DMDISFODS.The depth is indexed by h such that h = 0 represents the bottom of 3D matrix and h = 1 mm the top.
In total, 37 images were captured for a 1-mm scan, although only 9 images are shown in Fig. 4(a) for image presentation. Figure 4(b) shows the images of fluorescent beads captured by DMD-ISFODS. For comparison, wide-field images are provided in Fig. 4(c). Wide-field microscopy here is based on the same optical set-up illustrated in Fig. 1, except that the DMD does not produce pinhole aperture arrays and all DMD pixels reflect light. As expected, the fluorescence images taken by multiple pinhole aperture-based DMD-ISFODS provide much more crisp images of the beads by filtering the out-of-focus component of scattered light, although out-of-focus fluorescence is not completely blocked because of a large DOF. The large DOF is entailed by the long working distance that is needed for visualizing a thick 3D matrix.
For quantitative comparison of multiple pinhole aperture-based DMD-ISFODS and wide-field microscopy, we have introduced image blur ratio of a single fluorescence microbead. Image blur ratio here is defined as the ratio of the number of pixels with non-zero intensity to the total number of pixels
that are analyzed. This ratio can represent the amount of in-focus component over out-of-focus blur and be an indicator of image clarity when the DOF is much larger than the pixel size. Under the definition, measured images are sharper with a smaller image blur ratio. In Fig. 4, we have analyzed 52 fluorescence microbead images, each occupying an area of 35 × 35 pixels. Image blur ratio was determined as 0.18± 0.04 for DMD-ISFODS and as 0.43 ± 0.16 for wide-field images. In other words, DMD-ISFODS provides improved images over wide-field microscopy by 2.5 times in terms of image clarity.
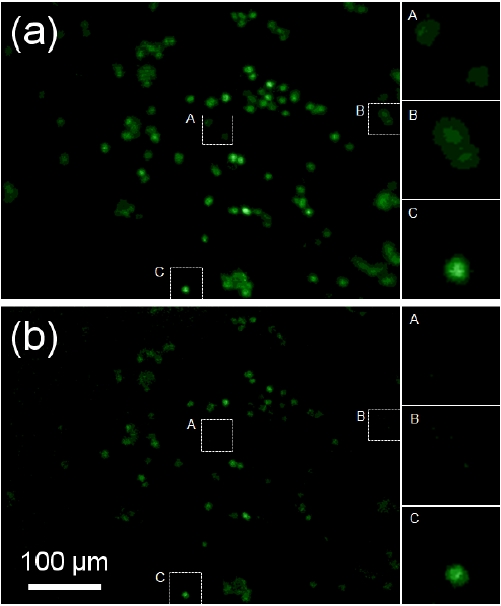
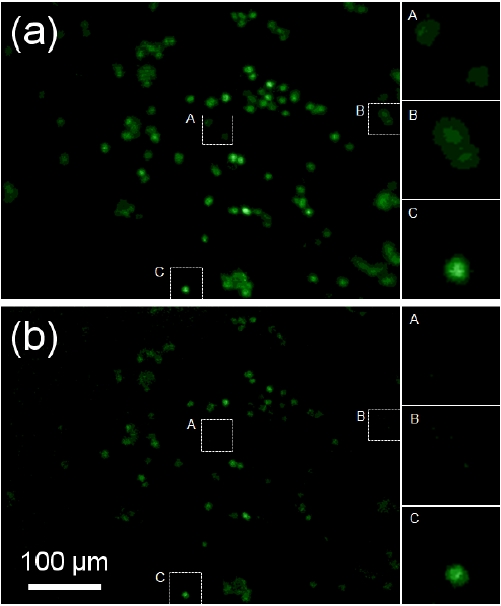
For imaging cells embedded in the 3D matrix, scanning was limited to a relatively short distance under 100 ㎛,because fluorescence emission efficiency of intracellular labels was low compared to that of fluorescent beads. However, similar to Fig. 4, improved images of cells were obtained with DMD-ISFODS as it removes background fluorescence more efficiently, as clearly shown when A and B are compared in Fig. 5(a) and (b). C in Fig. 5 also presents improvement of blurs in capturing cells in the focal plane. Regarding the limit in the scanning distance, we believe that the use of a more sensitive detector such as an EM-CCD, possibly combined with a coherent light source, would improve the efficiency and the scanning distance, although this would make DMD-ISFODS less portable.
The image clarity of DMD-ISFODS can further be improved, for example, by using structured illumination, which may require a high-power laser and a highly sensitive photo-imager because of low spatial filtering efficiency of a DMD [26]. Also, pinhole aperture scanning schemes may be improved: for example, modified scrambled-block Hadamard ensemble provides effective elimination of out-of-focused components [23].
In summary, we have designed and implemented a DMD-based fluorescence imaging system in situ for analyzing 3D cell cultures. The DMD-ISFODS was found to improve the clarity of fluorescence images over wide-field microscopy by 2.5 times by removing out-of-focus optical signals based on sophisticated scanning of multiple pinhole apertures. In contrast to optical detection systems developed previously, DMD-ISFODS can provide both lateral and axial information of thick biological samples over a very long working distance. The smallest depth resolution achieved was 6.3 ㎛ with a 100x objective lens. The feasibility was demonstrated over full 1-mm scanning through 3D alginate-based matrix embedded with fluorescent microbeads and A431 human carcinoma cells. This study suggests two directions of further research. First, both illumination and detection arms need multiple pinhole aperture scanning for additional improvement of image clarity. Also, complex multiple-layered microfluidic devices should be developed.