Lectins have the capacity to serve as recognition molecules between cells or organisms because of their noncatalytic sugar binding properties (Sharon 2008). Lectins are proteins that bind reversibly to carbohydrates, agglutinate cells, or precipitate polysaccharides and glycoproteins (Goldstein et al. 1980). Carbohydrate-binding properties of lectins have been applied in the fields of immunology, cell biology, cancer research, and genetic engineering (e.g., Sharon and Lis 2004).
Recently, the medical use of lectins has been studied extensively and some lectins exhibiting high anti-HIV (Sato et al. 2007) and antibiotic activities (Liao et al. 2003) were isolated. The use of lectins as pesticide agents (Wellman-Labadie et al. 2008) and potential drug delivery systems were also explored (e.g., Jung et al. 2010). Lectinbased cell-cell recognition systems have been proposed in many algal groups and in some cases the corresponding lectins have been isolated. However, the number of characterized lectins is still considered too small (Yoon et al. 2008, Han et al. 2011).
Marine green alga
For the successful aggregation of cell organelles during protoplast formation in
>
Organism and laboratory culture
The vegetative plants of
>
Preparation of concentrated algal extract and purification of BPL-4
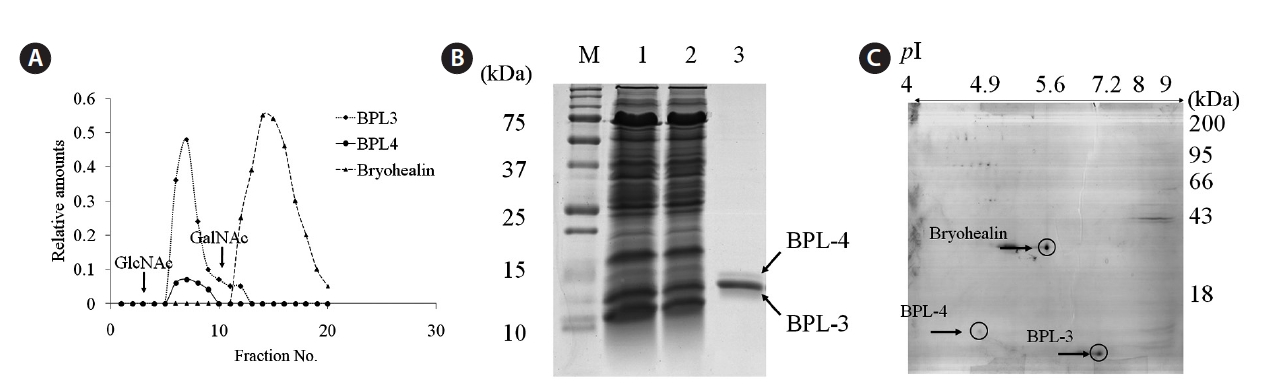
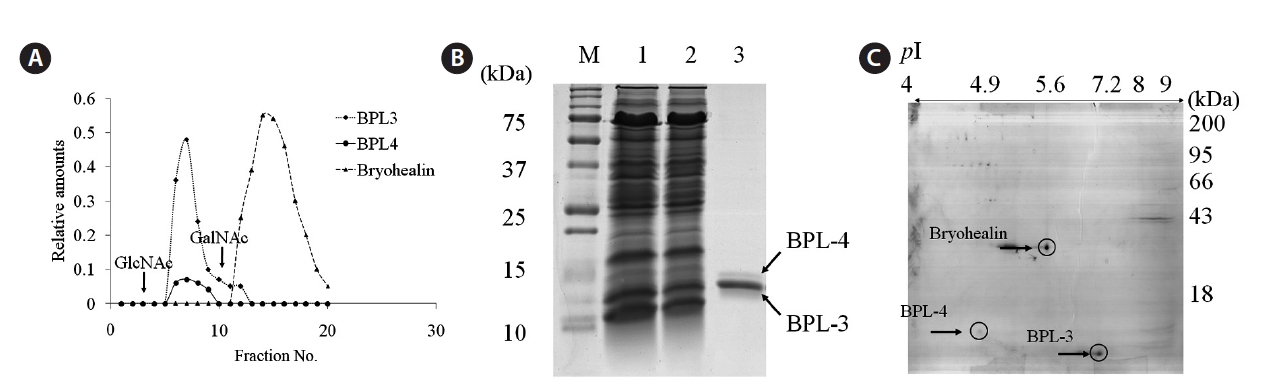
The concentrated algal extract was prepared as described by Han et al. (2011). It was loaded on N-acetyl-D-galactosamine (GalNAc)-agarose affinity column equilibrated with phosphate-buffered saline (PBS) of pH 7.3. The non-bound materials were removed with PBS until the absorbance at 280 nm was lowered to 0.001, and the bound protein was eluted with the same solution containing 0.05 M N-acetyl-D-glucosamine (GlcNAc). The fractions of 2 mL were collected and analyzed with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The fractions showing protein band and high hemagglutinating activity were pooled, concentrated, and dialyzed against PBS. The protein contents were determined by the method of Bradford (1976). SDS-PAGE was performed according to Laemmli (1970) with 15% gel. Samples were treated with 4% 2-mercaptoethanol to reduce disulfide bonds. Proteins were stained with 0.12% Coomassie brilliant blue R-250.
>
Two-dimensional gel electrophoresis (2-DE)
A mixture of elutes from GalNAc agarose affinity column was dialyzed in distilled water and lyophilized. Dried proteins were dissolved in 150 μL of rehydration buffer (7 M urea, 2 M thio-urea, 4% CHAPS, 2% ampholyte of pH 4-10) and centrifuged at 12,000 g for 15 min to remove insoluble materials. IPG dry strip (pH 4-10, 7 cm; Bio-Rad, Bedford, CA, USA) was rehydrated with 125 μL of sample in rehydration buffer for overnight at 20℃. Isoelectric focusing (IEF) was performed at 20℃ using a PROTEAN IEF Cell (Bio-Rad). The voltage was linearly increased from 250 to 4,000 V during 2 h, followed by constant 4,000 V with complete focusing after 10 kVh. After IEF, strip was incubated for 15 min in equilibration buffer (50 mM Tris-HCl at pH 6.8, containing 6 M urea, 2% SDS, and 30% glycerol), first with 1% dithiothreitol and second with 2.5% iodoacetamide. Equilibrated strip was inserted onto SDS-PAGE gel (7 × 7 cm, 15%) and run at 200 V, 20 mA constant. The gels were stained with Coomassie brilliant blue R-250 as described by Han et al. (2011).
>
Amino acid sequence analysis
The amino acid sequencing was carried out with an Applied Biosystems Precise Sequencer (Applied Biosystems, Foster City, CA, USA) in Korea Basic Science Institute (KBSI, Daejeon, Korea). The protein was electrophoresed on 15% SDS-PAGE and electroblotted onto a polyvinylidine fluoride membrane for 2 h at 4℃. Blotted membrane was stained with ponceau S staining solution. Internal amino acid sequence was obtained using chemically assisted fragmentation-matrix-assisted laser desorption / ionization (CAF-MALDI-TOF; Amersham Biosciences, Piscataway, NY, USA). Protein band was excised and digested with trypsin (Promega, Madison, WI, USA) and digested peptide was sent to Genomine Company (Pohang, Korea). The sonar in CAF-MALDI-TOF and MASCOT program was used for the identification of peptide sequence.
>
Construction of Bryopsis plumosa cDNA library
Double stranded cDNA was synthesized using 5 μg of poly (A) RNA as a template, directionally cloned into a UniZAP-XR vector phage (ZAP-cDNA synthesis Kit; Stratagene, La Jolla, CA, USA), and packaged using the ZAP-cDNA Gigapack III Gold packaging extract (Stratagene). Approximately 1.8 and 1.5 million recombinants were represented in the cDNA libraries, respectively.
>
RNA isolation and cDNA cloning
Complete BPL-4 cDNA was cloned in two steps. First, a degenerate forward primer [BPL4-NDF1, 5′-CA(A/G)
GC(A/T/G/C)GG(A/T/G/C)CTIGT(A/T/G/C)AA-3′] was designed from the amino acid sequence Gln-Ala-Gly-Leu-Val-Leu of a part of N-terminal sequence determined by Edman degradation and then a DNA fragment (~500 bp) was amplified from phage DNA with primers BPL4-NDF1 and T7 (in the lambda ZAP vector) under the following conditions: initial denaturation at 95℃ for 3 min, followed by 40 cycles of 94℃ for 30 s, 55℃ for 45 s, and 72℃ for 1 min, and then final extension at 72℃ for 10 min. This amplified DNA fragment was cloned into a QIAGEN PCR Cloning kit (Qiagen, Valencia, CA, USA) and sequenced. Second, specific reverse primer [BPL4-SF1, 5′-ATAGATATCAGGGTGTGCTG-3′] was designed according to the cDNA sequence and then another DNA fragment was amplified from the phage DNA with primers BPL4-SF1 and SK (in the lambda ZAP vector) under the same conditions as described above. The nucleotide sequences of both polymerase chain reaction (PCR) products were assembled and complete sequence of BPL-4 cDNA was obtained.
>
Computational sequence analysis
Nucleotide and amino acid sequence homology searches and comparison were carried out using BLAST on GenBank (http://www.ncbi.nlm.nih.gov/genbank), EMBL (http://www.ebi.ac.uk/embl), PDB (http://www.rcsb.org/databases.html), and Uniprot (http://www.uniprot.org). Post-translational modification of protein was identified using the CBS prediction server (http://www.cbs.dtu.dk/services). Secondary structure was predicted using PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred). Multiple alignment of protein sequence was performed using BioEdit program (ver. 7.0.9.0) with BLOSUM62 matrix.
We used two-step elution method to purify BPL-4 because
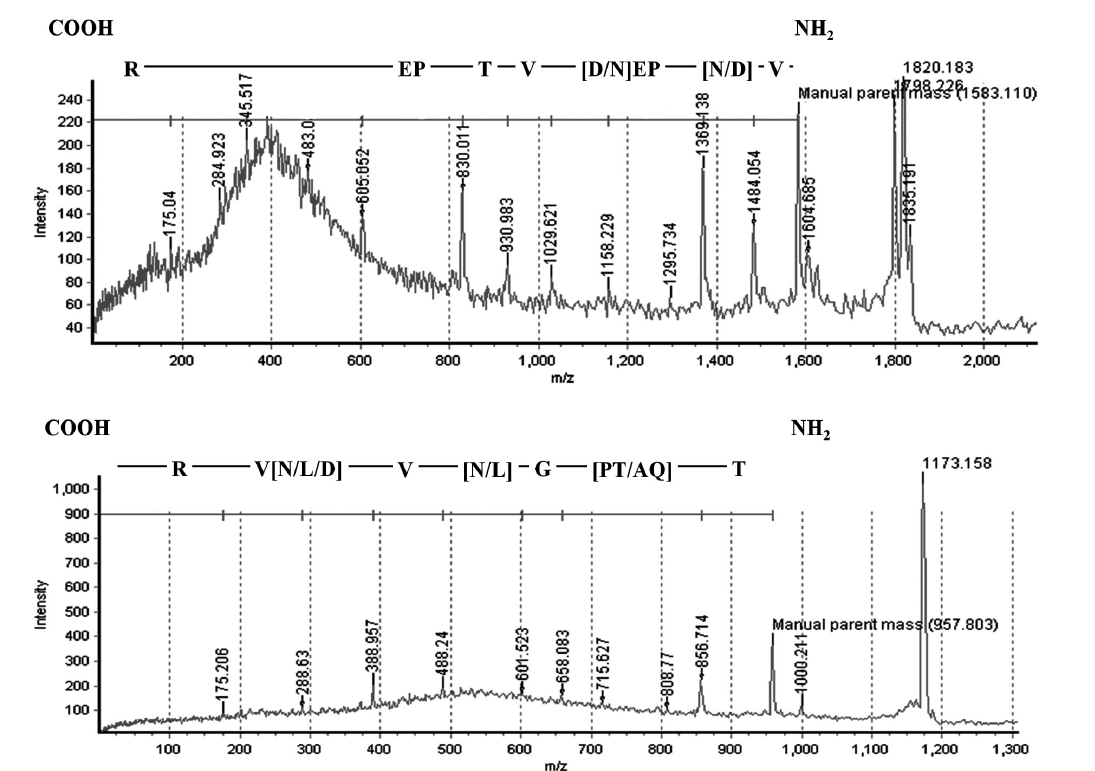
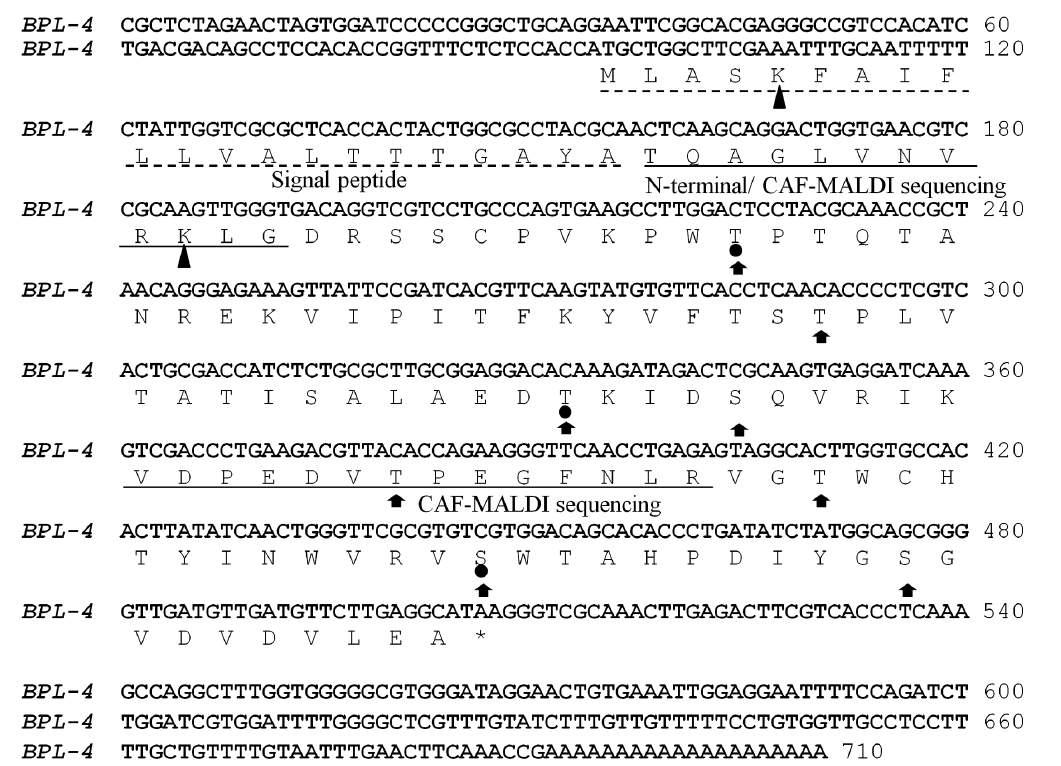
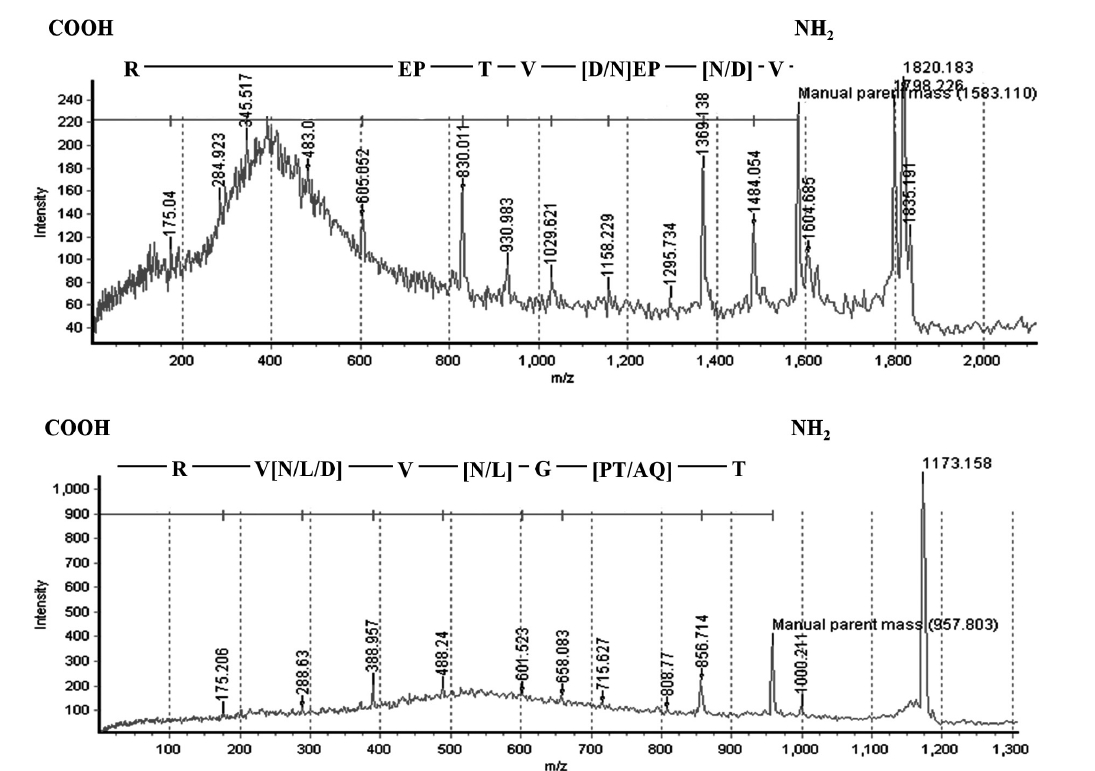
The N-terminal amino acid sequence of BPL-4 was determined using Edman degradation method; Thr-Gln-Ala-Gly-Leu-Val-Asn-Val-Arg-Gly (BPL4-NT1, Fig. 3). To obtain more sequence information, the lectin was cleaved with trypsin and the resulting fragments were subjected to CAF-MALDI sequencing. The unique sequences Val-[Asp or Asn]-Pro-Glu-[Asp or Asn]-Val-Thr-Pro-Glu-[unknown]-Arg and Thr-[Gln-Ala or Pro-Thr]-Gly-[Asp or Asn or Leu]-Val-[Asp or Asn or Leu]-Val-Arg (Figs 2 & 3) were obtained. The parent masses of isolated peptides were 1,583 and 957 Da, respectively (Fig. 2). The degen-
erate primers for further molecular characterization of BPL-4 were designed from above amino acid sequences.
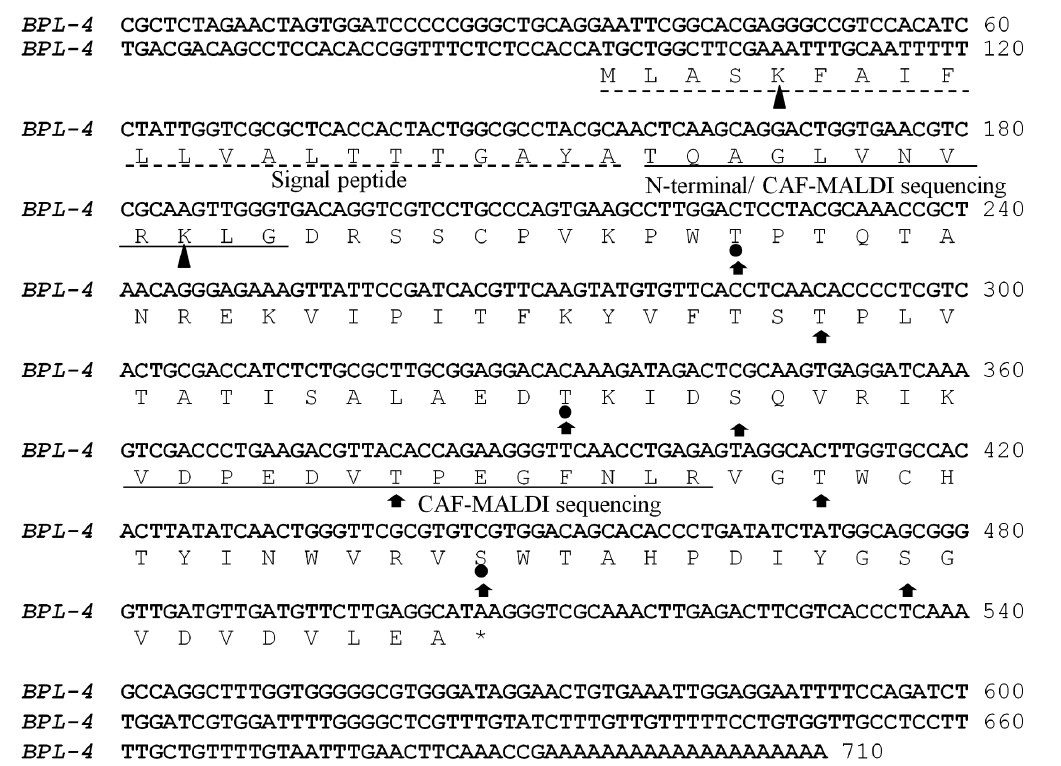
Full cDNA sequence (710 bp) of BPL-4 was obtained using PCR method based on the degenerate primers of the N-terminal amino acid sequence and specific primers from partial cDNA sequence. The BPL-4 cDNA consisted of 414 bp of open reading frame (ORF) and 203 bp / 93 bp of 3′ / 5′ untranslated regions. The deduced amino acid sequence of BPL-4 contained two known internal tryptic peptide sequences and N-terminal amino acid sequence obtained from the protein. The calculated mass of tryptic digested peptides agreed well with CAF-MALDI data (Figs 2 & 3).
>
Molecular properties of BPL-4
The protein consisted of 137 amino acids including N-terminal signal peptide (Fig. 3). Molecular weight without the signal peptide was calculated to 12.9 kDa and the isoelectric point was predicted to p
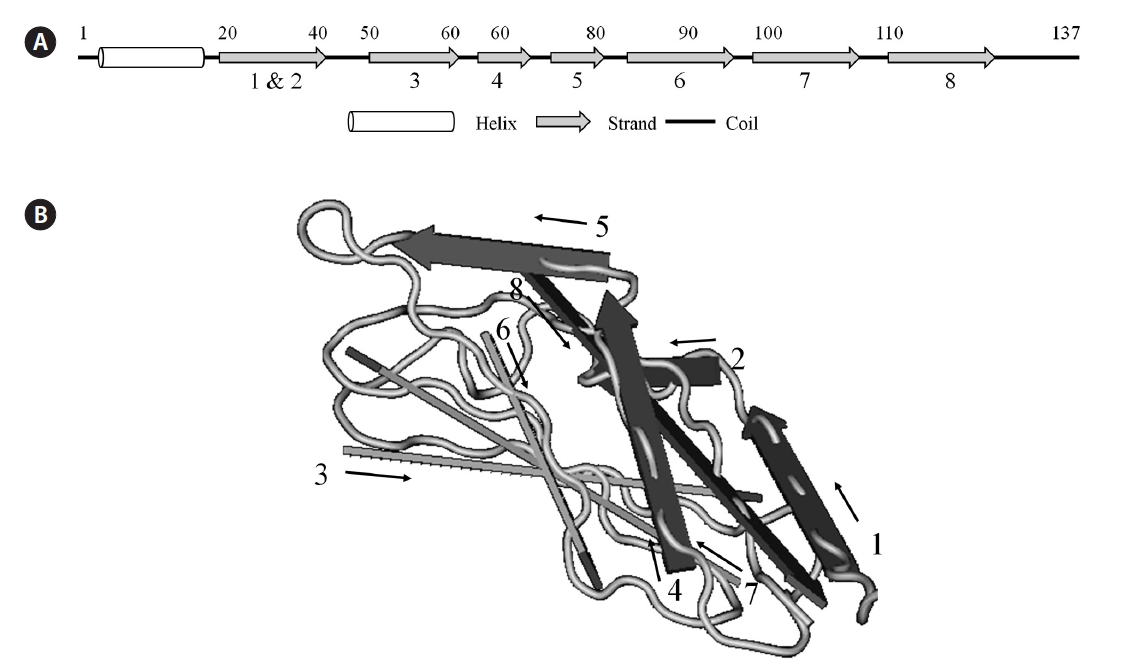
Two possible glycate sites (Lys5 and Lys31; score, 0.854 and 0.745) were predicted by the calculation with web-based prediction program (CBS Prediction Servers, http://www.cbs.dtu.dk/services), but the weak potential value (<0.593) of BPL-4 suggested that there was no glycosylation site. During the analysis of Yin-Yang and phosphotylation sites, 3 possible Yin-Yang sites (Thr44, Thr80, and Ser118) and 8 phosphorylation sites (Thr44, Thr66, Thr80, Ser84, Thr96, Thr106, Ser118, and Ser128) were found in BPL-4 sequence (Fig. 3). Analysis of secondary structure of BPL-4 using PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred) showed α-helix structure at the N-terminus and following eight ß-stranded wall structures. The predicted secondary structure of BPL-4 conformed well to the tertiary structure of the subunit of H lectin (
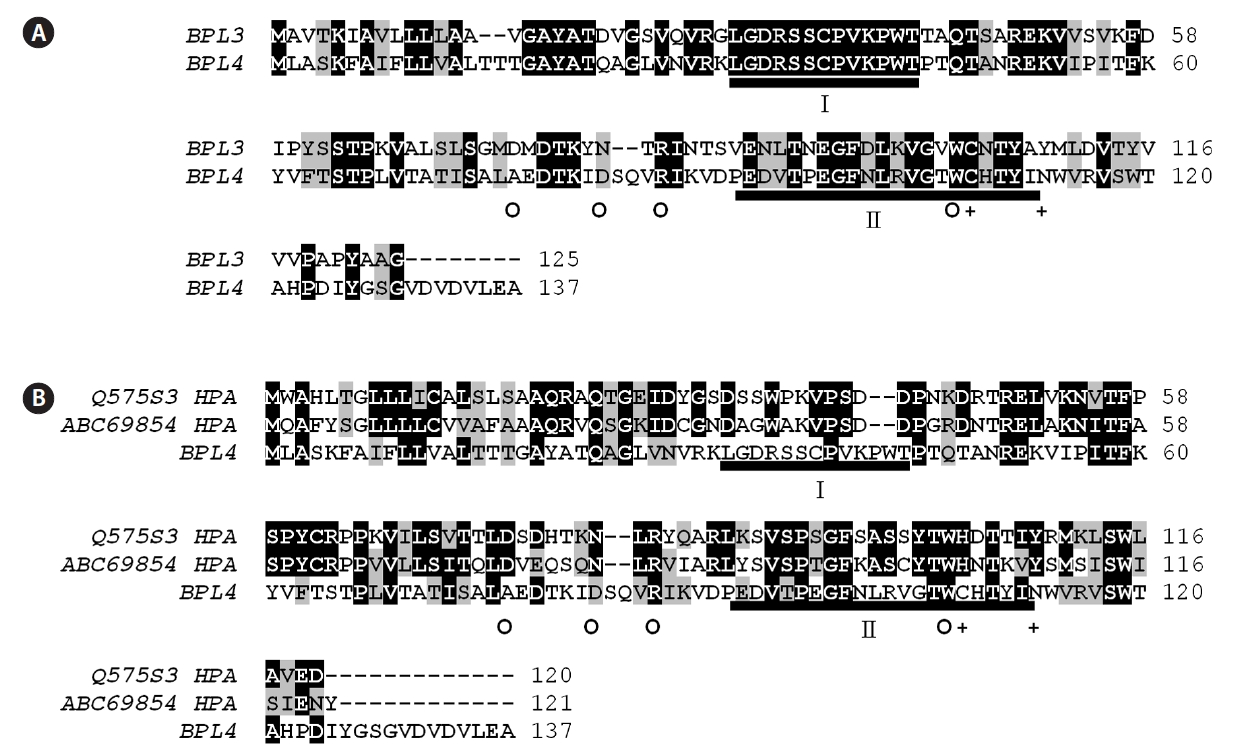
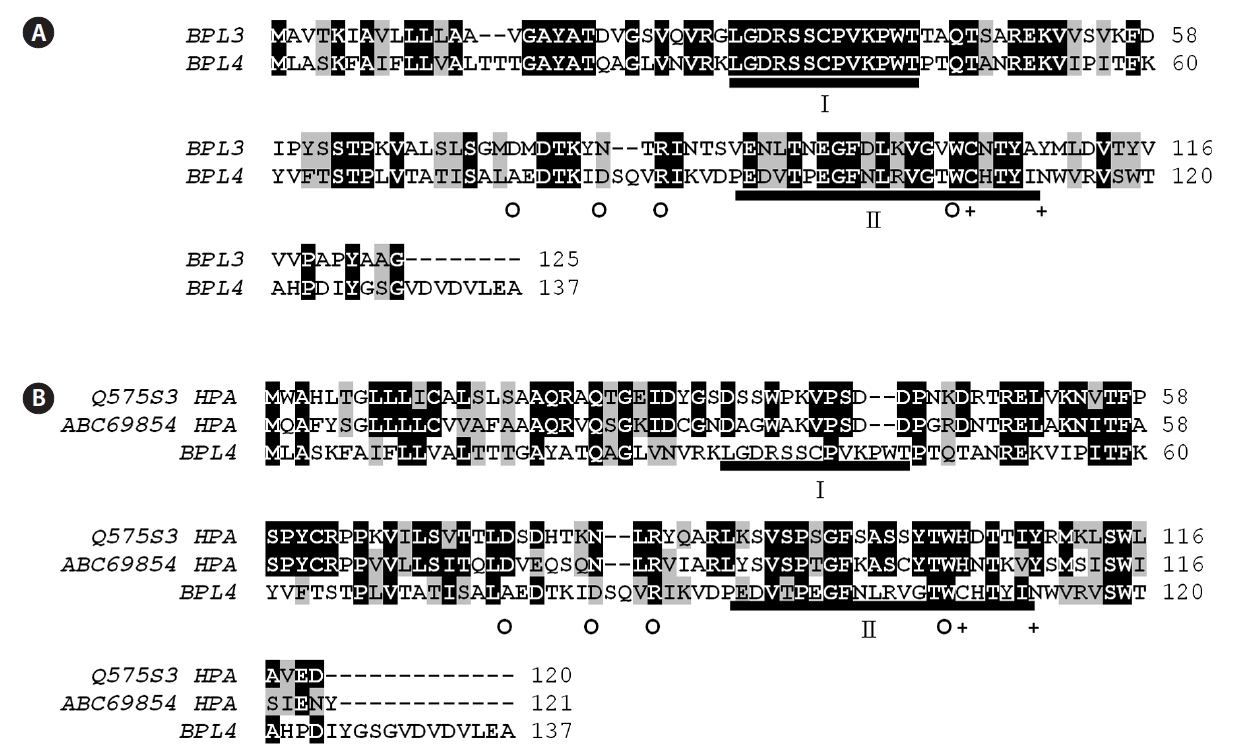
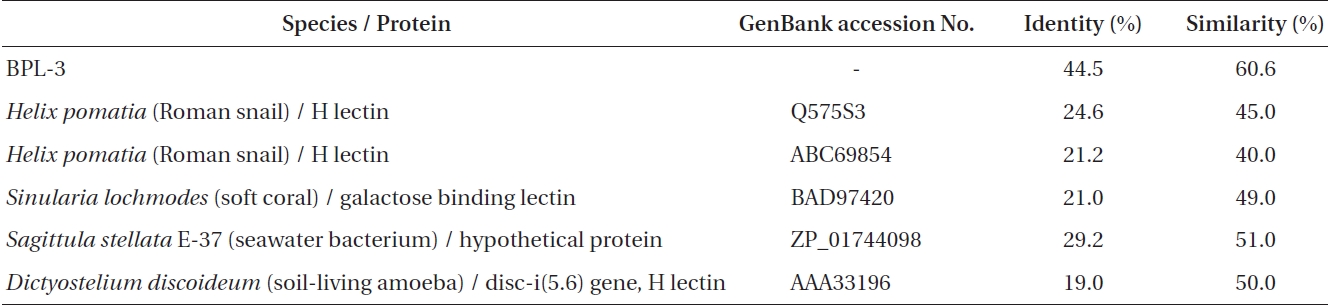
BPL-4 showed significant similarity with BPL-3, but it was clearly a different protein (Table 1, Fig. 5). BPL-3 and BPL-4 shared two well conserved domains including putative active domain of H lectin group, which is also an GalNAc and D-galactose binding lectin (Fig. 5). The similarity values ranged from 40 to 51% with other H lectins.
The amino acids involved in hydrogen bonding through their side chain were identical between
Possible involvement of lectins has been suggested for the protoplast regeneration of the marine coenocytic green alga
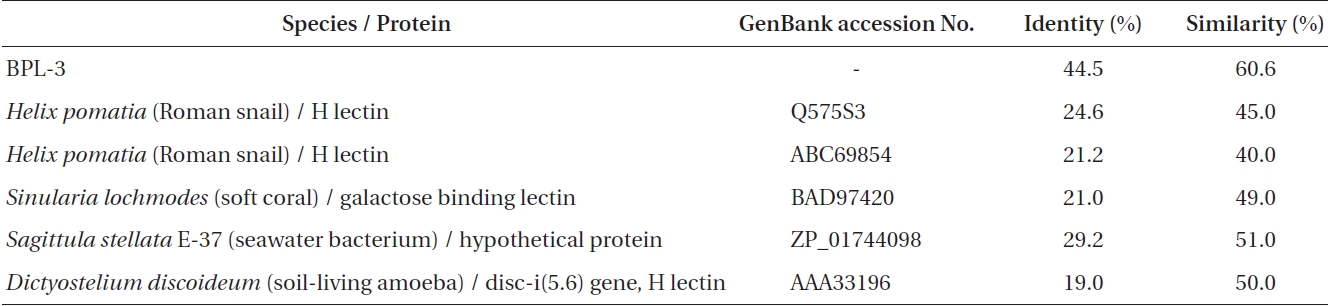
[Table 1.] Sequence identity and similarity between BPL-4 and related proteins
Sequence identity and similarity between BPL-4 and related proteins
posed for the aggregation mechanism of the extruded cell organelles in seawater (Kim et al. 2006). So far, three lectins (Bryohealin, BPL-2, and BPL-3) have been isolated in this species (Yoon et al. 2008, Han et al. 2010, 2011). The presence of additional lectin(s) specific to the sugars, GalNAc and GlcNAc was suggested because 1) the aggregation of cell organelles occurred at 2 different optimum pH values and 2) some residual lectin activity was remained in the column after purification of Bryohealin and BPL-3 (Han et al. 2011).
In this study, we purified another lectin, BPL-4, possibly involved in the protoplast formation of
Although BPL-3 and BPL-4 showed the same sugar specificity and similar primary structure they are clearly different lectins. The sequence similarity of two lectins was just 60.6% which was slightly higher than that (50%) between BPL-4 and an H lectin from amoeba
The evolutionary origin of BPL-4 is very interesting. Although BPL-3 and BPL-4 share many molecular characteristics in common, the analysis of their primary structure suggested that two lectins might have evolved separately. BPL-4 does not belong to any category of the terrestrial plant lectins just like BPL-3. But these two lectins show similarity with H lectins, which appear in octocoral and invertebrate animals and play a role in immunity and can bind to GalNAc and D-galactose (Koike et al. 2004, Jimbo et al. 2005, Sanchez et al. 2006). H lectins have been used as a research tool in the various fields of biochemical and medical sciences, as marker for tumor cells, for the antimicrobial and antiviral activities and hormone binding ability (Osborne and Brooks 2006, Sanchez et al. 2006, Bogoeva and Russev 2008). BPL-3 and BPL-4 are different from H lectin group in having slightly different sugar-binding specificity: H lectin show specificity to GalNAc and D-galactose, but BPL-3 and BPL-4 bind to GalNAc and GlcNAc not to D-galactose. The difference of amino acids in binding domains of these lectins may explain the difference of sugar binding specificity and hemagglutinating activity between BPL-3 and other H type lectins (Han et al. 2011).
Accumulating evidences show that algal lectins are involved in many biological processes, including wound response and sexual reproduction (Maier and Muller 1986, Kim et al. 1995, Ross et al. 2005, Yoon et al. 2008). However, the evolutionary origin of algal lectins is still in question. The result from molecular studies on algal lectins failed to show any direct relationship between algal and higher plant lectins (Yoon et al. 2008, Han et al. 2011). Instead, three lectins isolated from

![Analysis of secondary structure of BPL-4 using PSIPRED prediction program (http://bioinf.cs.ucl.ac.uk/psipred). (A) Predicted secondary structure of BPL-4. (B) Tertiary structure of the subunit of H lectin (Conserved domain database [CDD] number, pfam09458). The number and position of ß-strand wall structure of BPL-4 match with those of H lectin.](http://oak.go.kr/repository/journal/11253/JORHBK_2012_v27n1_55_f004.jpg)