In this study, novel
>
A. Bacteria Strains and DNA Preparations
Clinical isolates used in this study were provided by the Korean Institute of Tuberculosis and Department of Clinical Pathology, Seoul National University Hospital. Mycobacterial DNA samples were prepared by the bead beater-phenol extraction method [5].
A set of primers, which was previously used to amplify
>
C. DNA Hybridization in a Microtiter Well Plate
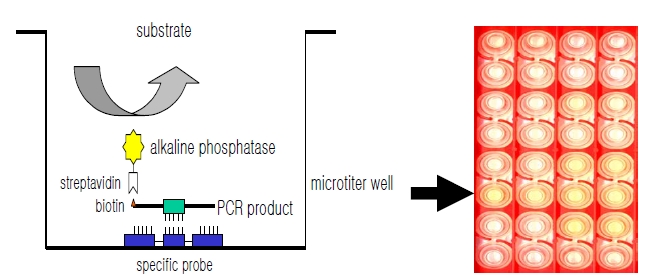
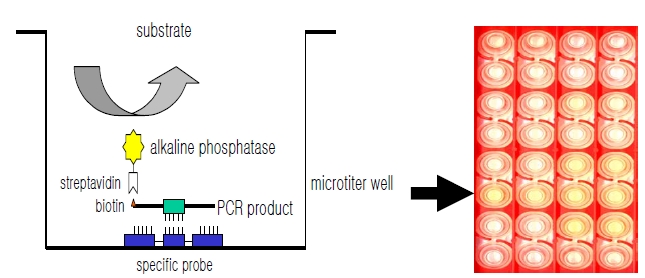
DNA hybridization was performed as previously described [6], with minor modification (Fig. 1). Briefly, a oligonucleotide-specific probe, kanp (5’-GCC-AGC-TCTCCC- AGT-TCA-3’) was designed from the known
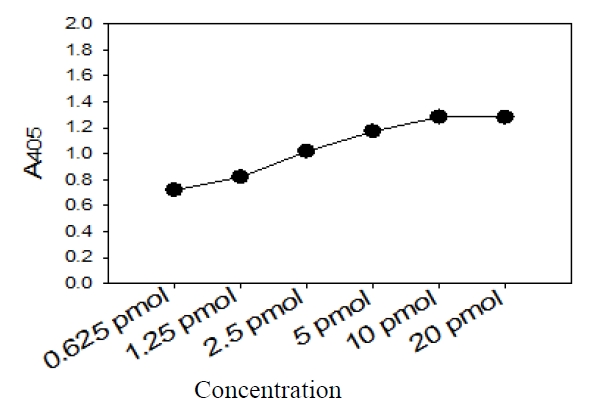
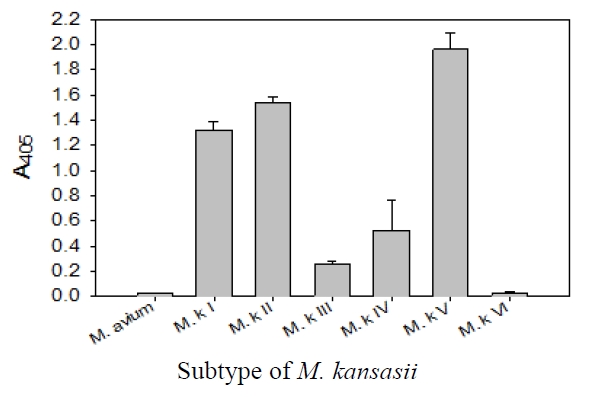
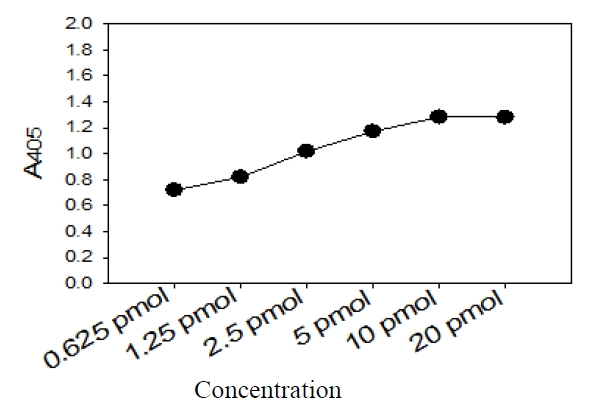
Five picomoles of the probe DNAs was dissolved in 50 μL of immobilization buffer (1.5 M NaCl, 0.3 M Tris?HCl [pH 8.0], 0.3 M MgCl2) and then dispensed into a microtiter well (NucleoLink Strips; Nunc, Rochester, NY, USA) and incubated over night at 37℃. The probe DNA mixture was then removed from the well and dried at 37℃ for 30 min. Wells were UV irradiated at 254 nm, for 5 minutes using an electronic UV crosslinker CEX-800 (Ultra-Lum, Claremont, CA, USA) and washed three times with 200 μL of washing buffer (1 M NaCl, 0.1 M Tris?HCl [pH 9.3], 2 mM MgCl2, 0.1% Tween 20). Probe-coated wells was immediately stored at 4℃ and used for hybridization. One hundred microliters of hybridization solution (5 × SSC, 5 × Denhardt’s, 0.2% SDS, and 200 μg/mL of salmon sperm DNA) was dispensed into a probe-coated micro-titer well. Five microliters of the heat-denatured PCR product was mixed with the hybridization solution and incubated in the well for 30 minutes at 50℃. The mixture was then removed from the well, which was rinsed three times with 200 μL of 2 × SSC. One hundred microliters of alkaline phosphataseconjugated streptavidin solution (Amersham Life Science, Arlington Heights, IL, USA), diluted 1:2000 with incubation solution (0.3 M NaCl, 0.1 M Tris?HCl [pH 7.5] 2 mM MgCl2, 0.05% Triton X-100), was then added to the well, and incubated for 15 minutes at room temperature. After incubation, the well was rinsed three times with 200 μL of incubation solution, then 100 μL of 1 M diethanolamine buffer (pH 9.8) containing 0.5 mM MgCl2 and 10 mM p-nitrophenyl phosphate was added, and the whole solution was kept at room temperature for 60 min. The enzyme reaction was stopped using 5 μL of 10 M NaOH and the optical density at 405 nm (OD405) of each well was read using a micro-titer plate reader (Multiskan Ascent; Labsystems, Grand Rapids, OH, USA). The OD405 values of triplicated wells were use to draw a bar graph using SigmaPlot 2000 (Ver. 6.00) (SPSS Inc, Chicago, IL, USA).
The nucleotide sequences of the purified PCR products were directly determined as previously described [1].
For the sequencing reaction, 60 ng of PCR amplified DNAs, which were purified using a QIAEX II gel extraction kit (QIAGEN, Hilden, Germany), 5 pmol of either the forward or the reverse primer, and 4 μL of BigDye Terminator v2.0 100 RR mix (Perkin-Elmer Applied Biosystems, Foster City, CA, USA) were mixed, and the contents were adjusted to a final volume of 10 μL with distilled water. The reaction was run for 30 cycles of 10 seconds at 96℃, 5 seconds at 60℃, and 4 minutes at 60℃. Both strands were sequenced as a crosscheck. Determined sequences were compared with those of reference strains in GenBank to compare sequence similarities.
This specific oligonucleotide probe, kanp probe, for
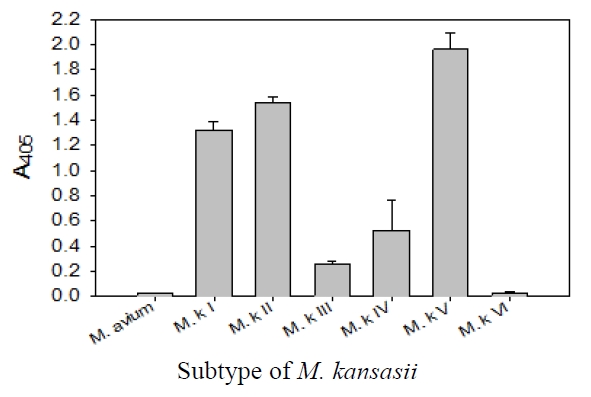
This probe also was proved to be specific in terms of the amplification of the
This probe was specific for subtype I, II, IV, and V of
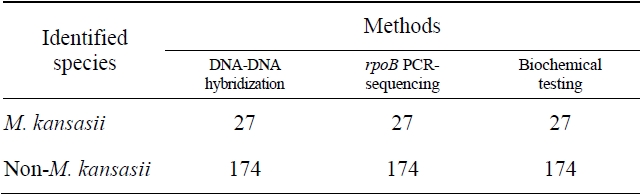
DNA-DNA hybridization test for detection and identification of
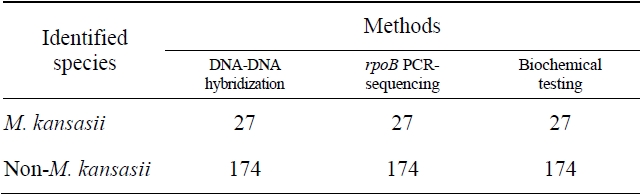
Comparision of the results obtained by DNA-DNA hybridization method and other methods for identification of M. kansasii
The sensitivity and specificity of this DNA hybridization method performed with culture samples was 100%. The negative strains by this DNA hybridization method were identified as
There have been developed many diagnostic methods for mycobacterial infections containing conventional culture methods, PCR-based methods, and liquid culturebased mycobacterial detection systems, such the Bactec [7], MGIT [8], ESP [9], and BacT/Alert 3D [10]. There are many PCR-linked methods using 16S rRNA gene [11-14] and 16S?23S rRNA spacer region [3, 14] in mycobacteria. However, despite these efforts, a good standard protocol for detection and identification of
In this study, we suggest that this DNA-DNA hybridization method assay using