Polyacrylonitrile (PAN)-based carbon fiber has been studied for decades [1,2]. Its excellent properties, such as high tensile strength, low weight, and low thermal expansion, make it very popular in aerospace, civil engineering, military, and motors ports, as well as in other competition sports [3,4]. The quality of precursor materials largely affects the final performance of carbon fibers [5], as does the thermal stabilization [6]. Earlier research on producing carbon fiber is always focused on how to improve the mechanical properties of the precursor fibers and reducing the induction period and rate of heat release during stabilization [7]. However, previous studies are rarely involved in the stereoregularity of PAN configuration, which is beneficial for cyclization during stabilization [8]. Since there are many nearby nitrile groups in high isotacticity polymer groups, this increases the cyclization degree paired with low tacticity. The high isotacticity of the precursor materials results in higher crystallinity and mechanical properties, so the final carbon fibers’ properties may improve by increasing the isotacticity of precursor fibers.
Nowadays, high stereoregularity of PAN can be achieved by acrylonitrile-urea canal polymerization initiated by γ-ray irradiation [9] and matrix-anionic-polymerization [10, 11]. In this study, high tacticity PAN, with tritacticity of 0.53, was successfully synthesized by anionic polymerization, in non-polar solvent, using dialkylmagnesium (R2Mg) as an initiator. The high stereoregularity iso-PAN’s crystallinity, used as a precursor, is effective in producing high performance carbon fiber. Moreover, the optimized pre-oxidation of iso-PAN is essential, which is seldom investigated in previous studies. As pre-oxidation is a very complex process with a variety of elementary reactions such as cyclization, oxidation, aromatization, dehydrogenation, etc., it can hardly be studied by using a single model [6, 12, 13]. Detailed study of the chemical reactions, structural evolution and kinetics during this process will provide the necessary theoretical basis for optimizing the process and performance improvement of the final carbon fiber.
In this study, the structural evolution of high isotacticity PAN isothermal-treated at 200, 220, 250, 280℃ was investigated by Fourier transform infrared spectroscopy (FTIR) and wide-angle X-ray diffraction (WXRD) respectively. Unreacted nitrile groups fractions were calculated by the peak intensity of nitrile groups at 2240 cm-1 and 1600 cm-1, which is the intensity of C=C and C=N. Besides, thermal properties of iso-PAN were analyzed by using differential scanning calorimetry (DSC). A kinetic study was constructed from DSC dates with different heating rates of 5, 10, 15, 20℃. Activation energy and pre-exponential factor were determined by iso-conventional methods, viz. Kissinger and Flynn-Wall-Ozawa (FWO) methods [14].
2.1. Synthesis of high tacticity PAN and isothermal treatment of iso-PAN
A 100 mL four-necked flask equipped with a stirrer, dropping funnel, condenser, two-way cock and thermometer was needed for polymerization. The flask was flushed with dry nitrogen for 5 min, 0.053 g (1,4-cyclohexanediol) and 40 mL of Xylene were added. After dissolving the alcohol completely at 130℃, the initiator, (n-C6H13) 2Mg an n-heptane solution, was added into the flask while stirring, causing white solvent-insoluble substances to form immediately. The solution was kept within the initiator at 138℃ for 15 min, then 5 mL of acrylonitrile (AN) were added dropwise while stirring. The polymerization started instantly and soon the solution became brown. The polymerizations were allowed for 7 h, then hydrogen chloride (HCl) and CH3OH solutions, as polymerization terminators, were added dropwise while stirring. After cooling to room temperature, the solution was collected by filtration through a glass filter (G3 grade), and successively washed with acetone and water, and thereafter dried in a vacuum for 24 h, allowing white PAN powder to be obtained.
Isothermal treatments at different temperatures (200, 220, 250 and 280℃) were conducted in a precision controlled tube furnace with a temperature accuracy of 1°C. After the temperature in the furnace reached the settle temperature, the iso-PAN was placed into the furnace and pre-oxidation started immediately. Samples with different reaction degrees were obtained at various times during the reaction time at 5, 10, 15, 30 and 60 min.
FTIR spectra of samples were recorded on a Nicolet-210 (Nicolet, USA) Spectrophotometer with a wave range of 4000-400 cm-1. Weighted samples were mixed thoroughly with a constant amount of KBr and palletized. As the reaction proceeded, the nitrile groups with an absorbance at 2240 cm-1, reacted to produce cyclized units containing ?C=N- and ?C=Cgroups,which absorbed at 1595 cm-1. Reaction degrees can be estimated via unreacted nitrile groups fractions by using the following formula:
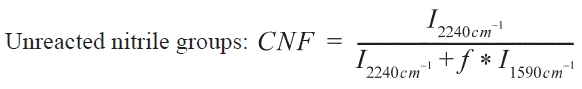
Where I2240cm-1 is the absorption of nitrile groups, I1595cm-1 is the absorption of ?C=C? and ?C=N? groups. f is the ratio of nitrile groups and cyclized groups absorptivity constants, f = 0.29 [15].
The XRD pattern of high isotacticity PAN with different cyclization degrees was recorded on a Rigaku D/MAX 2500 VB2+/PC X-ray diffractometer (Japan) operated at a voltage of 40 kv and a current of 30 mA with nickel-filtered Cu Kα radiation (λ = 1.54056 A), and a scanning speed of 5?/min range from 5-40?. Crystal size (Lc) of the laterally ordered phase can be acquired from the Scherrer equation:

Where K is the apparatus constant and β is the half-value width FWHM of the intensity versus the Bragg angle.
Crystallinity (C) can be calculated with the following formula:

Where χc is the area of the crystalline portion in the XRD patterns, χa is the area of the amorphous portion in the XRD patterns [7].
DSC measurements were carried out using a STA449C calorimeter (Netzsch, Germany). Iso-PAN samples with the mass of 4-7 mg were placed into the coil which was made of an aluminum-based alloy used for DSC measurements. The same empty coil was used as a reference. The specimen was heated in the instrument with different heating rate of 5, 10, 15, 20℃/min from 45 to 450℃ in an air atmosphere with a flow of 40 mL/min.
3.4. Characteristics of high tacticity PAN
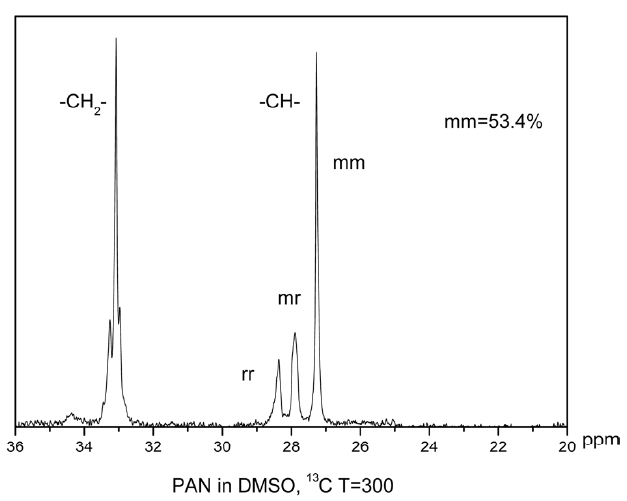
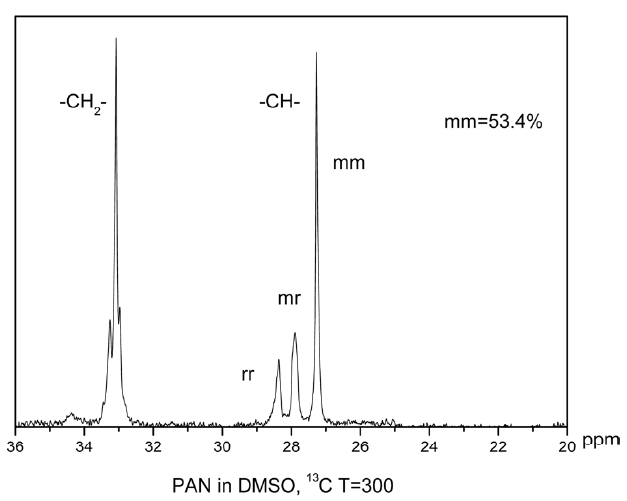
Nuclear magnetic resonance (NMR) analysis was carried out using an Av600 wide-bore solid state NMR spectrometer equipped with 3.2 mm HCN balun probe operating at 200.968 MHz for 13C. Spectra were collected using proton decoupling (30 ms; 74 KHz). During acquisition, pulse delay times were 1.745 s, acquisition times 0.655 s, pulse widths 5.5 us (45opulse). Accumulation was more than 3000 percent.
The triad tacticities [(mm), (mr) and (rr); “m” and “r” mean “meso” and “racemo diad” sequences, respectively] were determined on the basis of the Schaefer’s assignment [16,17] via 13C NMR spectroscopy spectra, from the ratio of intensities of the three methane carbon peaks (26.9 ppm for mm, 27.4 ppm
for mr, and 27.9 ppm for rr methane carbon) in the 13C NMR spectrum.
By calculating the mathematical area of the three peaks, the tacticity of the polymer can be obtained: 53.4% (mm), 19.47% (mr), 27.13% (rr). The higher the value of mm is, the more nitrile groups exist in the same plane, which greatly affects thermal stabilization of the polymer.
4.1. Cyclization and oxidation of iso-PAN in an air atmosphere
Pre-oxidation of iso-PAN is a complicated process, involving cyclization, oxidation and some cracking reactions. Structural evolution in the process of isothermal pre-oxidation can be examined by FTIR.
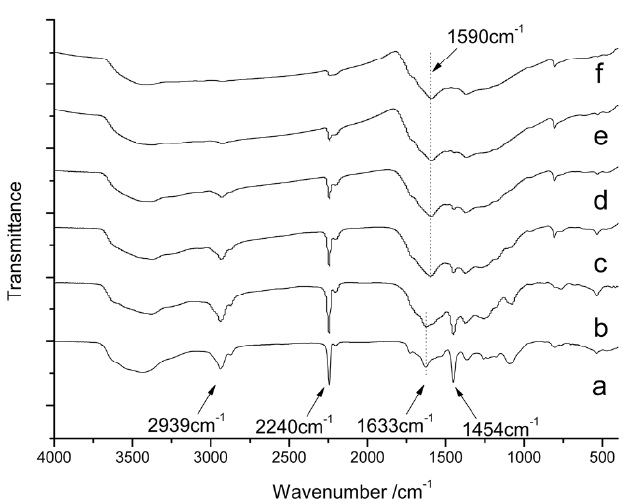
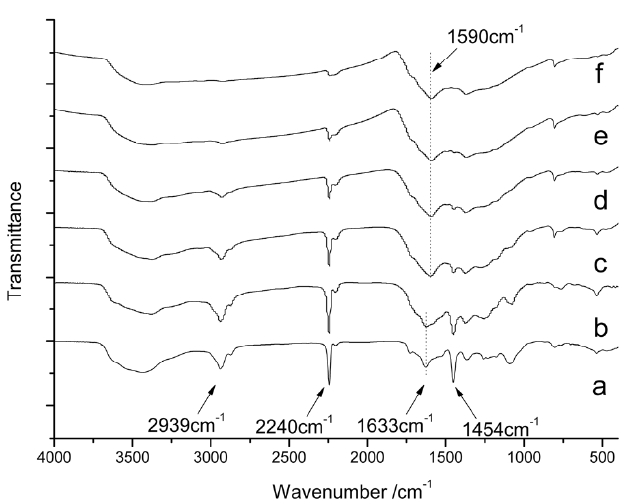
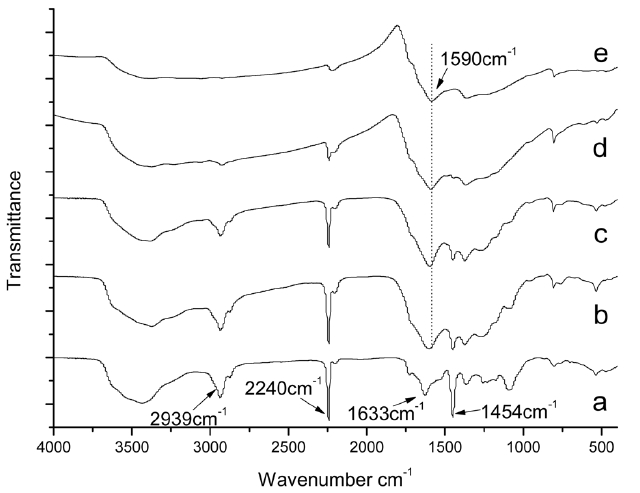
Fig. 2 shows the FTIR spectra of isothermal treated iso-PAN for various times at 250℃, among which Fig.2 a shows the spectra of the iso-PAN without heat treatment. The band at 2243 cm-1 corresponds to C-N stretching of the acrylonitrile unit in the polymer chain and the absorption bands at 2939 and 1454 cm-1 can be attributed to CH2 stretching and CH bending, respectively. The assignments of some other absorption bands are given as follows: 1369 cm-1 (bending of CH), 1227 cm-1 (twisting vibration of ?CH2). The weak absorption at 1632 cm-1 which is attributed to C=C vibration, is probably due to termination by disproportionation, which can be confirmed by the discoloration of diluted bromine water [18]. The wide absorption range between 4310-3520 cm-1 corresponds to the vibration of -OH of unsaturated water. By comparing the spectrum of heat treated PAN for different times (5, 10, 15, 30 and 60 min), it is seen that with the extension of heat-treated time, obvious decreases happen to the ?CN stretching band (2240 cm-1), which almost disappears over the long-treated time, as do the -CH2 stretching and -CH bending bands at 2939 and 1454 cm-1, respectively. The absorption bands appearing at 1590 cm-1 were characteristic of C=C and C=N combinations vibration ladder structures. As
shown in Fig.2 c, a wide shoulder band appears at 1730 cm-1 due to C=O being assigned to the stretching ketone, due to oxygen uptake. The changes of spectra from (a) to (f) indicated that nitrile groups are converted into cyclized C=N groups and cyclization, dehydrogenation, and even oxidation started progressing to a great extent as pre-oxidation proceeded.
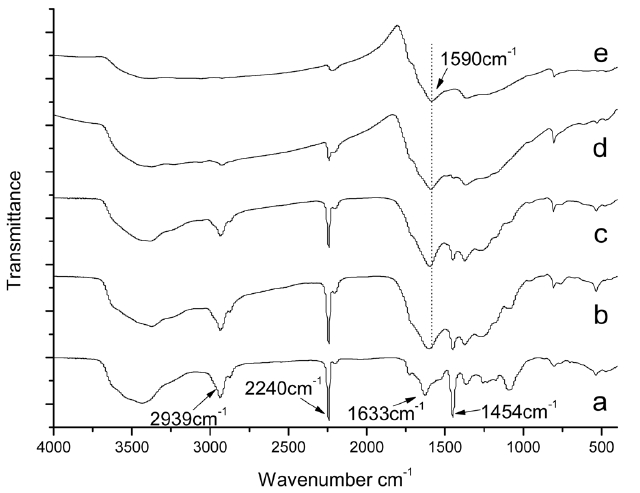
Fig. 3 shows changes of the FTIR spectra for isothermal treated iso-PAN at different temperatures. Fig.3 a presents the original spectrum of iso-PAN before heating. There is a significant decrease in the bands of nitrile groups (2240 cm-1). The absorption of the nitrile groups is clear and strong in Fig.3b, but indistinguishable. By Fig.3 e, it has almost disappeared,which implies that at a lower temperature, more nitrile groups remain, combined with absorption at the band of C=C and C=N, it can be concluded that heat-treatment is a crucial factor that affects structural evolution. The less residual nitrile groups are left over, the more liner polymer chains are converted into
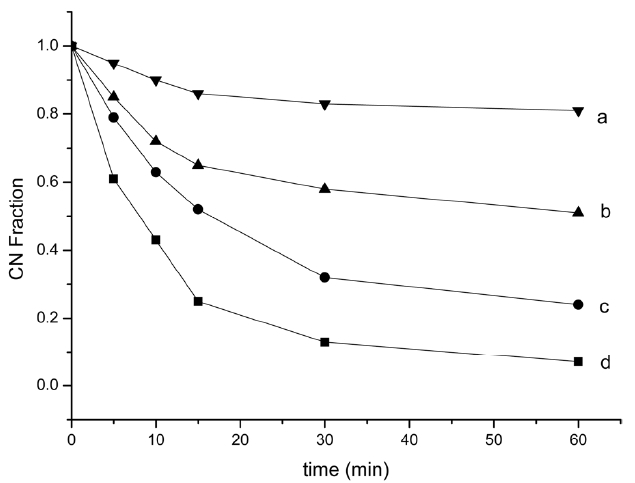
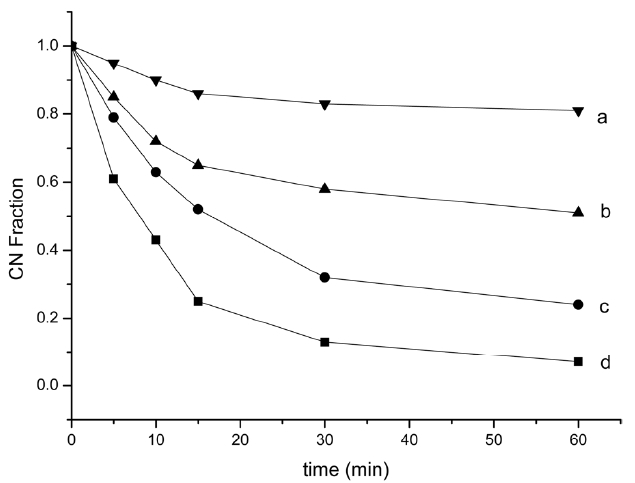
cyclized structures as temperature increases. According to the formula (1), residual CN fractions were calculated (Fig. 4), which can measure the extent of cyclization, using the intensity at 2240 and 1590 cm-1.
Fig. 4 shows the residual nitrile groups fraction (reduced carbon nanofiber [RCNF]) for different treatment temperatures and times. All four curves followed the same trend: RCNF decreased with increasing pyrolysis temperature and time. Violent cyclization reaction exhibited an acceleratory period for the first 15 min, where the reaction proceeded very rapidly. Most of the liner nitrile groups transformed into a cyclized structure and then entered a decay period where the reaction rate slowed down in the next 15 min, a steric hindrance effect of the cyclized structure. When time increased, the reaction entered into a retention period, when approximately 15%, 40%, 70% and 90% of all nitrile groups had reacted, at 200, 220, 250 and 270℃ respectively, at a very slow rate. The persistence of unreacted nitrile groups decreased when the heat increased, which may be attributed to the formation of isolated nitrile groups which were difficult to convert into a ladder polymer structure.
4.2. XRD analysis of isothermal treated iso-PAN
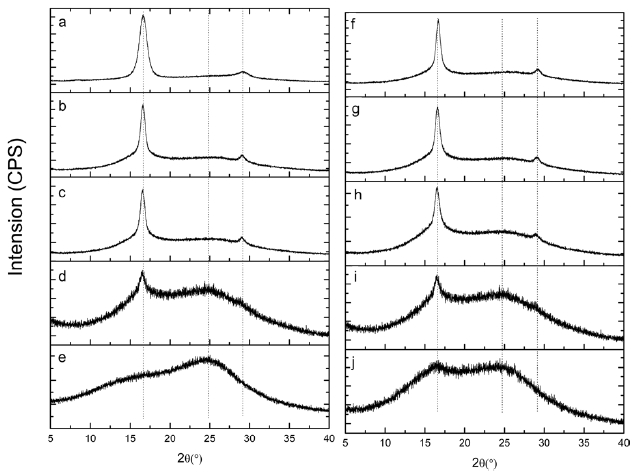
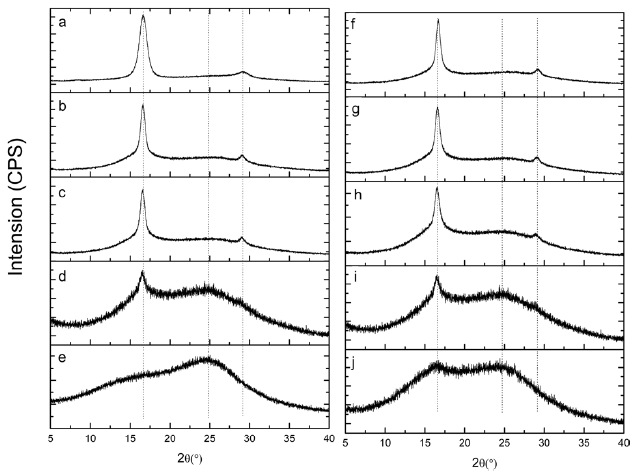
Fig. 5 shows XRD patterns of isotactic-PAN under different heat treatment temperatures for 30 min. A typical diffractions at 2θ = 16.5 and 29° in Fig.5 a, (100) and (110) planes, corresponds to the hexagonal unit of the C=N groups of the intramolecular dipole-dipole interactions among the nitrile group pairs and second-order hexagonal structure, respectively. As the stabilization goes, the intensity of the (100) peak became weaker and weaker with increasing temperature, and the (110) peaks disappeared gradually, when the temperature increased to 250℃. There is a broader peak centered approximately at 2θ= 25?, (200) plane, related to the irregular sheet-like structure of the ladder polymer, swamps the secondary peak at 29?, as shown in Figs.5 b-d. Stabilized at 280℃ for 30 min (Fig.5 e), the diffraction of (100) at 2θ = 16.5? is difficult to distinguish. This could be due to the destruction of the regularity of iso-PAN when the linear C≡N in the hexagonal domain transformed into
a new ladder structure. The higher the temperature was, the higher the cyclization degree of the iso-PAN.
Figs.5 f-j shows X-ray diffraction patterns of isotactic-PAN under 250°C in an air atmosphere for different times. The typical diffractions at 2θ = 16.5? and 2θ = 29? corresponding to (100) and (110) planes became weaker and weaker, but a little change and minor decrease in the first 15 min, (Figs.5 f-h), implies that a few nitrile groups of linear ordered structure were destroyed and a cyclization reaction hardly occurred in the regular domain. As shown in Fig.5 i, the intensity of (100) decreases significantly, and the intensity of (110) disappears, which indicate that the regularity of iso-PAN was destroyed and most of the linear nitrile groups were converted into ladder structures and new regular structures formed which caused the intensity at 2θ = 25? to become stronger. When prolonging the reaction time to 60 min (Fig.5 j), the intensity at 2θ = 16.5? continues to decrease, and, on the contrary, the intensity at 2θ = 25? increases as the reaction proceeds, indicating more and more linear structures turned into ladder structures and new regular structures were formed.
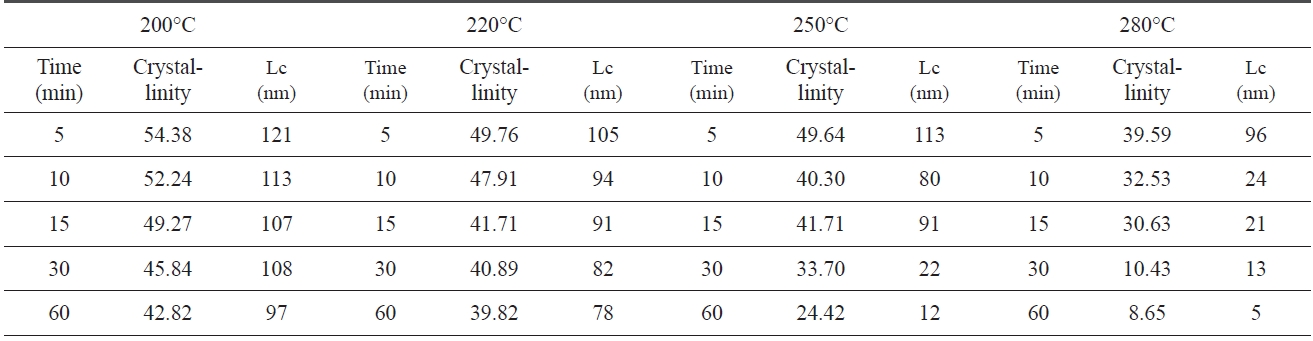
An XRD of isothermal treated samples was measured. Of each sample, crystallinity and crystal size which were calculated via Eqs. (1-2) and the Scherren formula, as shown in Table .1 At 200℃, the crystallinity and crystal size are at 54.38% and 121 nm at 5 min., respectively. However, the crystallinity and size decreased to 42.82% and 97 nm at 60 min., as the pre-oxidation proceeded during the isothermal treatment, likely, at higher treat temperatures (280℃). The crystallinity and crystal size of PAN decreased from 39.59% and 96 nm to 8.65% and 5 nm. These results suggest that the longer the heat treatment of PAN, the lower the crystallinity and size, and with a decrease in speed of crystallinity and size there is an increase in pre-oxidation temperature.
Crystallinity and crystal size which are closely related to the regularity of a PAN polymer chain, decreased as pre-oxidation proceeded, because linear polymer chains converted into ladder structures during isothermal heat treatment.
[Table 1.] Crystalline parameters of iso-PAN during iso-thermal treatment
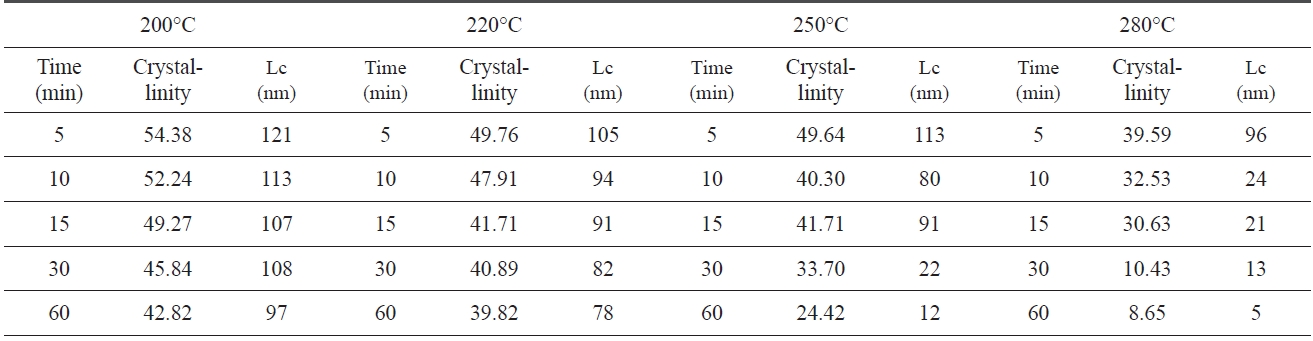
Crystalline parameters of iso-PAN during iso-thermal treatment
4.3. Evaluation of kinetic parameters of iso-PAN via DSC curves
There are some methods to evaluate activation energy of pre-oxidation by the value of the cyclization index and aromatization through FTIR and XRD [8,15] by isothermal treatment of iso-PAN. However, there are some defects in each method which can only evaluate elementary reactions during pre-oxidation. However, such methods are not suitable for pre-oxidation containing elementary reactions. The iso-conversional methods involving the FWO [19] and Kissinger-Akahira-Sunose (KAS) methods [20], were used for analysis of kinetic pre-oxidation by means of DSC dates of iso-PAN at different heating rates [21].
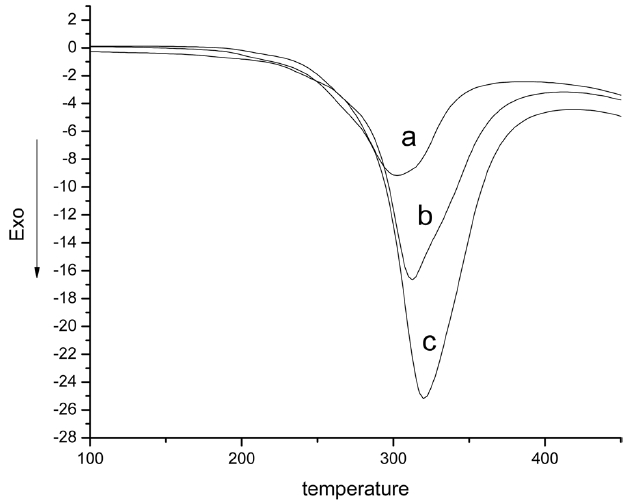
Fig. 6 shows DSC curves of iso-PAN at different heating rates. The cyclization reaction combined with oxidation with different heating rates resulted in strong exothermic peaks in DSC analysis. The exothermic peaks wholly shifted to a higher temperature. The peak temperatures were 302.5, 312, and 319.8℃ respectively, and the exothermic peaks became stronger and stronger. This is because the polymer has had enough time to fully react. The thermal effects and thermal inertia of the reaction increased with an increase in heating temperature. Samples could not be heated in time, so the cyclizations only took place at higher temperatures and the exothermic peaks shifted to a higher temperature. DSC analyses were performed at different temperatures. The conversion (α) at any time (t) was obtained from the relation:

Where Ht is the fractional heat of the reaction at t min and ΔH is the total enthalpy.
The rate of conversion is the linear function of the temper-ature-dependent rate constant, k(T), and the temperature-inde-pendent function of conversion, f(α).

Where t(s) is time and T(K) is the absolute temperature, k(T) can be described by the Arrhenius equation:

Where A (s-1) is pre-exponential and E (Kj mol-1) is the activation energy, R (Kj mol-1K-1) is the universal gas constant.The function f(α)is expressed as:

Where n is the reaction order, so the formula 3-6 can be modified to:
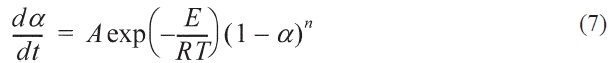
The heating rate Φ can be described as
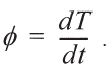
Eq. (7)could be transferred to:
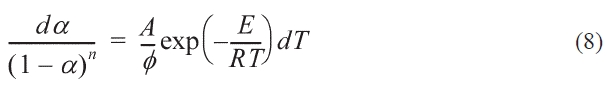
The integration function of Eqs. (8) is shown as:
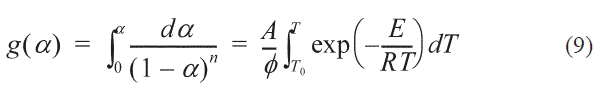
Where T0 is the starting temperature, the KAS, FWO formulas can be obtained by transforming Eq. (9) into Eqs. (10, 11):
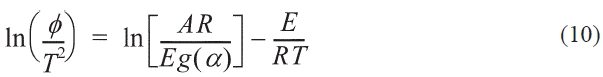
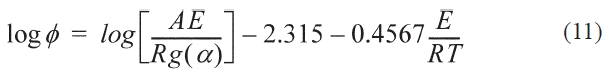
The advantage of the two methods is activation energy can be evaluated without knowing the reaction model and mechanism of pre-oxidation. So, the kinetic parameters can be evaluated by these methods without assuming a reaction model but rather the peak temperatures of DSC analysis at different heating rates.
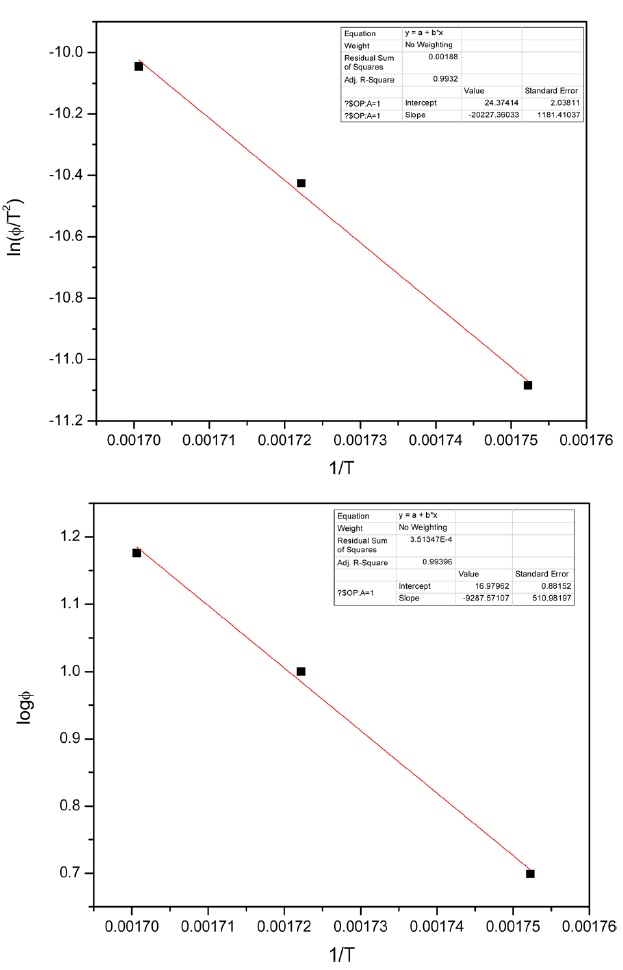
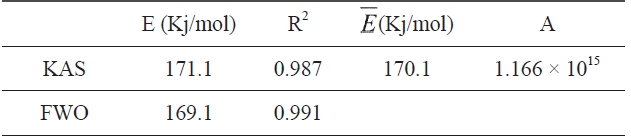
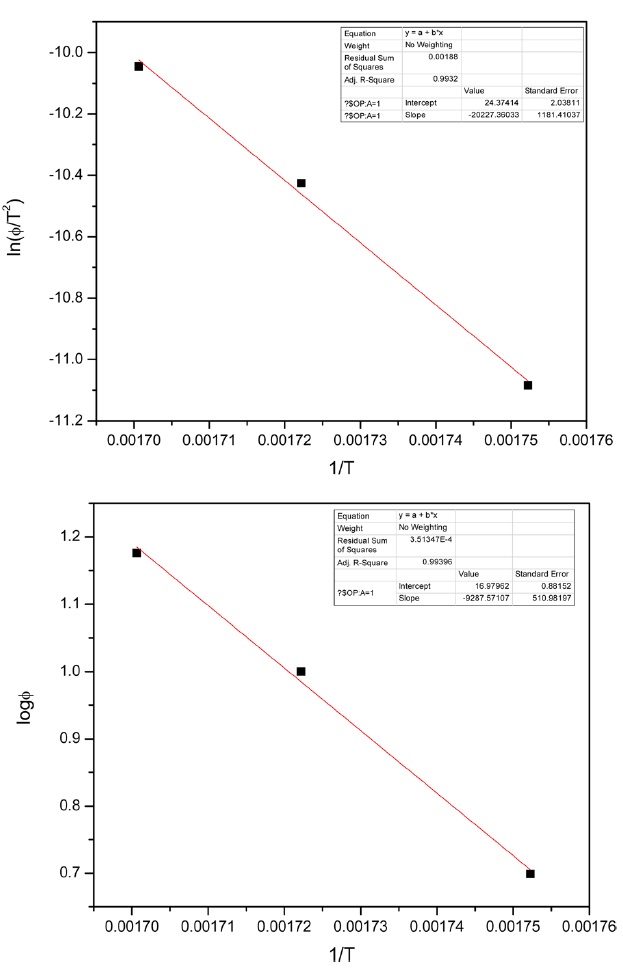
Kinetic parameters of pre-oxidation by iso-conversional methods, KAS and FWO according to Eqs. (10) and (11), plots of ln Φ, ln(Φ/T2) against 1/T should be straight lines, as shown in Fig. 7 Activation energies can be evaluated from the slope of the linear plots by the two methods. Pre-exponential factor A
[Table 2.] Kinetic parameters determined by KAS and FWO methods
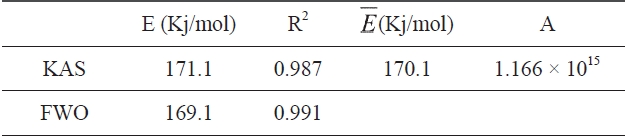
Kinetic parameters determined by KAS and FWO methods
can be calculated in both cases from the following formula [22]:
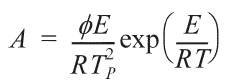
The activation energies and pre-exponential factors of the reaction are listed in Table 2:
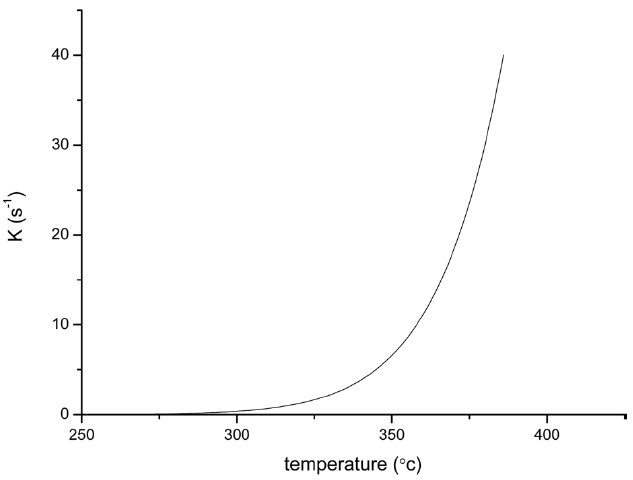
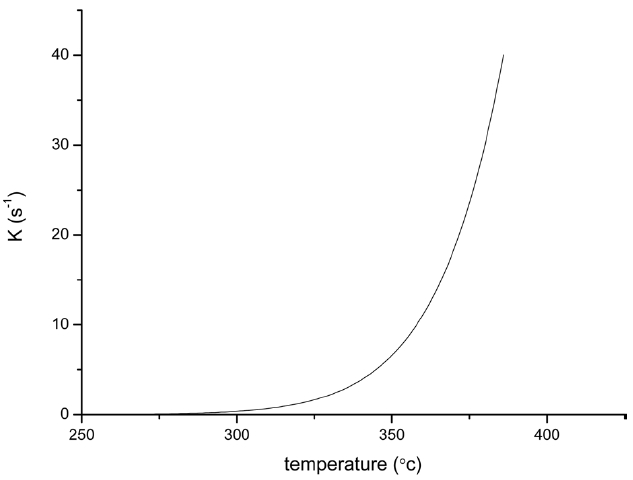
From the calculated data in Table 2, Ea and A values by KAS and FWO methods give nearly identical values, which indicate that the chosen iso-conversional methods are reasonable. So, the sensitivity of the pre-oxidation reaction during heat treatment can be correctly reflected by the parameters. The iso-PAN has a higher activation energy which implies that iso-PAN is more sensitive than at-PAN, via comparing the previous study to the current one [8]. The higher stereoregularity of iso-PAN makes more nitrile groups exist in the same plane, which can lower the cyclization reaction temperature and speed up the reaction rate. The rate constant k at different temperatures can be calculated from the Ea and A by the Arrhenius equation and is plotted in Fig. 8 It can be found that the rate of the cyclization reaction of iso-PAN increases with temperature. According the results, a centered release of heat during stabilization can be avoided by controlling the heat treatment temperature.
Isotacticity PAN with triad isotacticity of 0.53, using R2Mg as an initiator was successfully synthesized. Structural evolution and chemical changes during the isothermal treatment were investigated by FTIR and WXRD analysis. The residual nitrile groups fractions decreased with increasing temperature, and crystallinity and crystal size decreased proportionally to the pyrolysis temperature. The kinetic parameters of the reaction were determined by iso-conversion methods, and the rate constant was calculated. The results indicate that iso-PAN is highly sensitive to treatment temperature. This study gives scientific support for controlling stabilization temperatures which is beneficial for producing high performance carbon fibers.