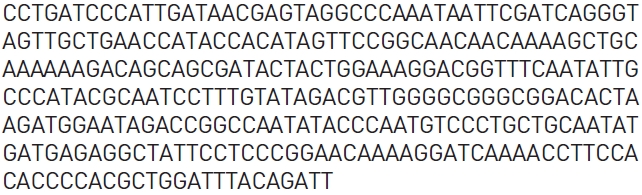Ginseng,
Cultivated ginseng is cultivated artificially and accounts for the majority of ginseng in the current market. Mountain wild ginseng grows in natural environments, vegetating in deep mountains, while mountain cultivated wild ginseng is seeded and grown in forests and mountains and is considered as a mimicry of mountain wild ginseng. And they have been shown to contain higher levels of ginsenosides. On the other hand, the reported differences in total ginsenoside contents between wild and cultivated ginseng were minimal [6-7]. In both Korea and China, wild ginseng is widely accepted to be more active than cultivated ginseng in chemoprevention. However, because of its high cost and sparse distribution,few systematic studies on wild ginseng have been done, and little has actually been reported on the differences between wild ginseng and other types of ginseng. Also, the lack of quality control has led to chaos in market distribution [8-9]. Thus, our research team conducted a study to identify wild ginseng specific genes for standardization. We succeeded in identifying one novel clone, the
Light-induced photosynthetic water oxidation and plastoquinone reduction takes place in the thylakoids of plants. These redox-mediated reactions are catalyzed by a multi-subunit membrane complex designated as photosystem II [11]. The minimum subcomplex that can evolve oxygen and release protons is referred to as the PSII core complex. More than twenty five different protein subunits make up the photosystem II complex of oxygenic photosynthetic organisms [12].
At the heart of this complex is the reaction center consisting of the D1 and the D2 proteins, where primary charge separation occurs [13]. Closely associated with the D1 and the D2 proteins are two similar chlorophyll a-binding proteins, CP43 and CP47(product of the [14]. These proteins serve as an"inner antennae" system that is linked to a secondary light-harvesting system.
Here, we have cloned the
2.1. Various ginsengs for RNA isolation
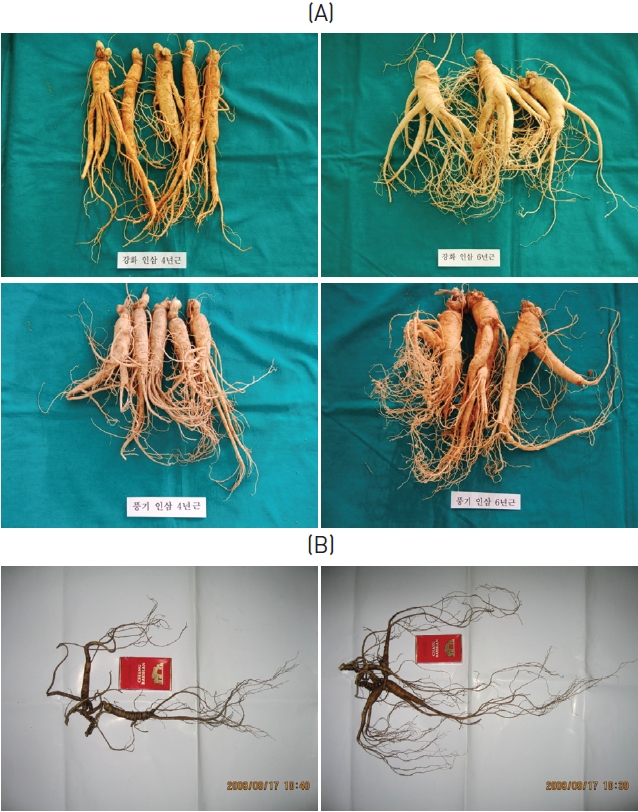
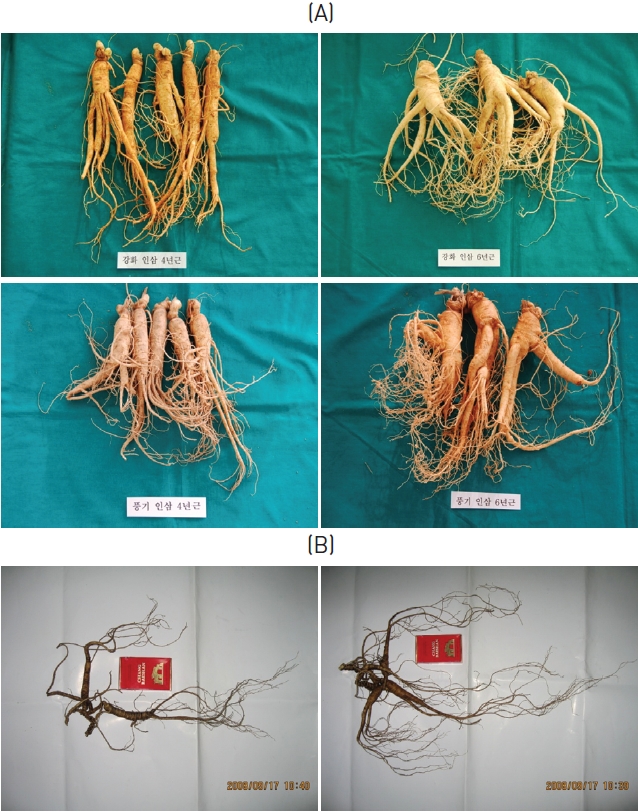
The cultivated ginsengs (CGs) used in this experiment were 4 and 6 years of age and from various region in Korea. The wild ginsengs (WGs) used in this experiment were collected from Changbai Mt. in 2008, and were about 20 to 40 cm long, with masses of about 20 to 30 g and approximate ages of 30 to 50 years (Fig.1 ).
2.2. Total RNA isolation and mRNA purification
Ginseng was ground in liquid nitrogen by using a mortar and pestle, and RNA was isolated using the RNeasy Plant RNA Isolation Kit (Qiagen). The concentration of isolated RNA was estimated by measuring its absorbance at 260 nm. An aliquot of the RNA extract was treated with DNase-I (Invitrogen) prior to cDNA synthesis by using Superscript III reverse transcriptase(Invitrogen) and random hexamers according to the manu-facturer|s protocol.
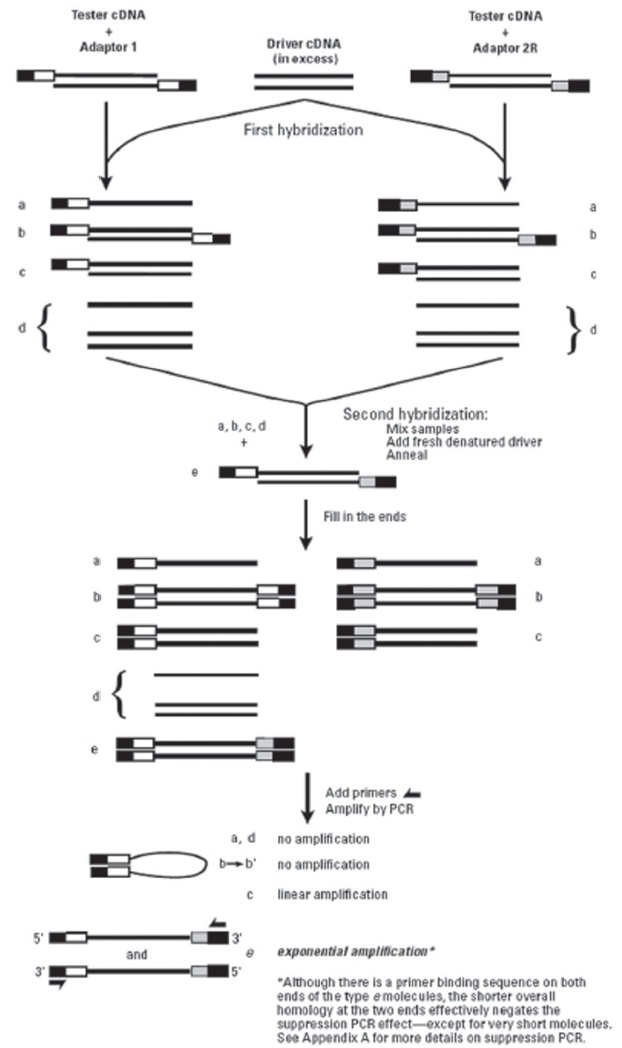
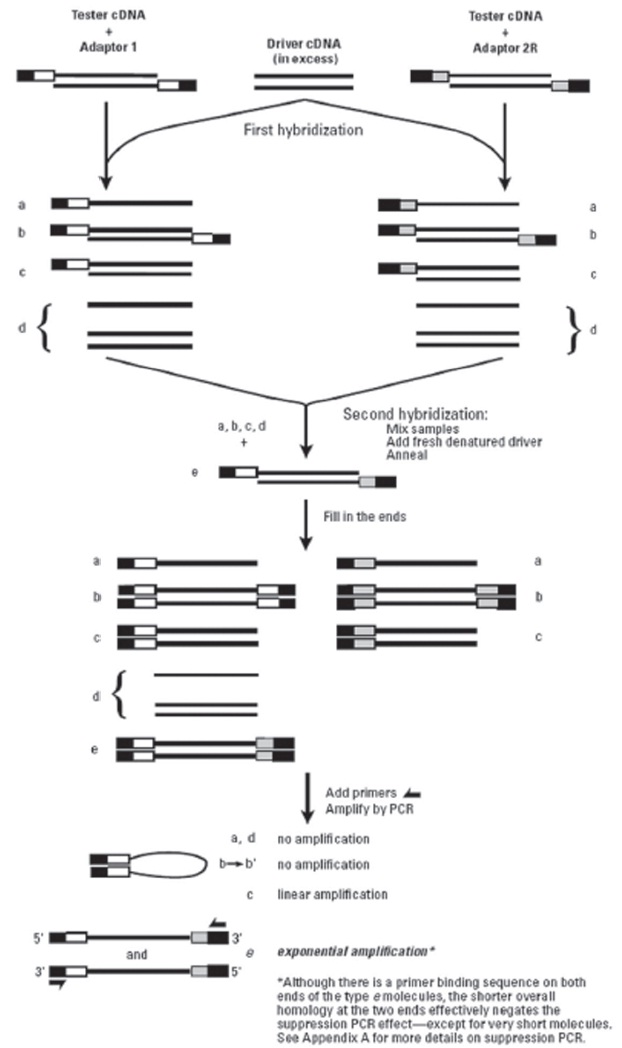
2.3 Suppressive subtractive hybridization
Suppressive subtractive hybridization (SSH) was performed using Clontech PCR-SelectTM cDNA Subtraction Kit (Clontech)according to the manufacturer’s protocol. SSH method includes several steps (cDNA synthesis, RsaI digestion and adaptor ligation, two rounds of hybridization and PCR) for isolating differentially expressed genes.
The cDNA fragments, derived from SSH forward subtractive library (tester: mountain cultivated wild ginseng; driver:ginseng), were cloned into pEC-T vector (KOMA Co., Seoul, Korea). The positive clones containing inserted fragments were identified by using the colony-PCR method.
Semi-quantitative RT-PCR was performed to compare the differential expression of the genes in the SSH library by using gene-specific primers. Total RNA (2 μg) was used for cDNA synthesis according to the First Strand cDNA Synthesis Kit(Invitrogen), and 1.0 μl of cDNAs was used as a template for PCR. PCR amplification was performed under the following conditions: 95°C for 5 min, 30 cycles at 95°C for 45 s, 54°C for 30 s, and 72°C for 60 s. The final incubation was done at 72°C for 5 min. PCR products were electrophoresed in a 2% agarose gel.
2.5. Quantitative Real-time quantitative RT-PCR
Real-time quantitative RT-PCR detection was performed with a StepOne machine and Fast SYBR Green Master Mix (Applied Biosystem, USA) and were measured in a 96-well plate. For each well, the 20 μl reaction involved 10 μl of the 2 X Fast SYBR Green Master Mix, 0.5 μM each of forward and reverse primer, 2.75 μl of DNase-free H2O, 2 μl of cDNA templates. PCR reactions were performed using the following parameters: 8 min at 95°C and 40 cycles of 45 s at 95°C, 45 s at 56°C and 45 s at 72°C. PCR products were melted by gradually increasing the temperature from 60°C to 95°C in 0.5°C steps.
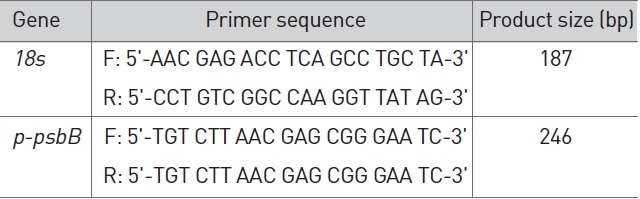
The identities of the amplicons and the specificity of the reaction were verified by using a melting curve analysis. Normalization of the cDNA templates was achieved by using 18S quantification. The primers presented in Table 1 were used to analyze
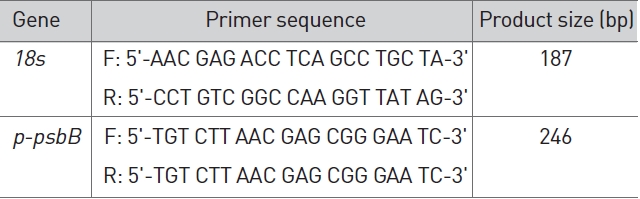
Primer for RT-PCR.
2.6. Sequencing and homology analysis
PCR products were cloned into the pEC-T vector (KOMA Co., Ltd, Seoul, Korea) and then sequenced by using the ABI 3700 DNA sequencers (PerkinElmer Applied Biosystems). The sequence analysis was performed using Chromas sequence analysis software. BLASTn was used to study similar nucleotide sequences.
3.1. Isolation of differentially expressed genes in wild ginseng
To identify wild ginseng-specific genes, wild ginseng cDNAs were subtracted from a pool of cultivated-ginseng cDNAs (Fig.1). The subtraction was expected to significantly reduce common cDNAs and to enrich for wild-ginseng-specific cDNAs.More than 100 transformants were obtained from the library, and the recombinant efficiency detected by using colony-PCR was about 90%.
One hundred positive clones confirmed by PCR amplification were randomly selected, from which, 16 significantly different clones were sequenced. Because the suppression subtractive hybridization procedure includes a restriction enzyme digestion of the cDNAs produced, none of the clones obtained from the resulting libraries were full length.
Among the novel cDNAs identified here as putative wild-ginseng-specific genes is a putative chloroplast
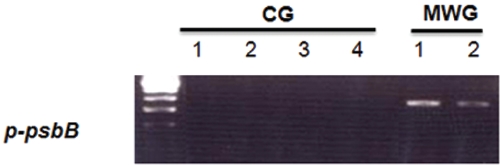
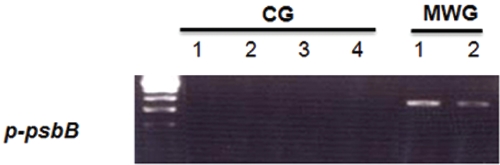
To confirm the differential expression of the
As shown in Fig. 4, all of the
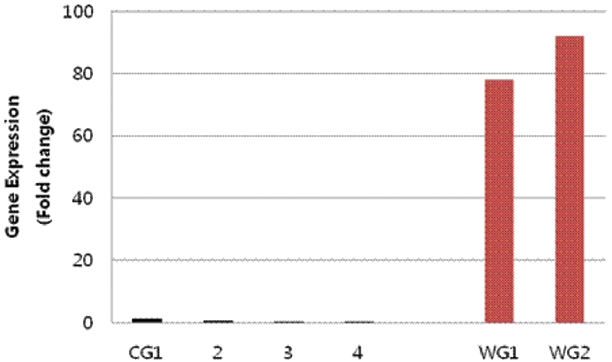
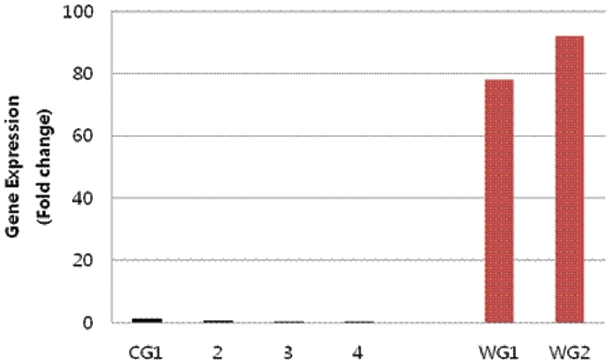
3.3. Real-time RT-PCR analysis
To further verify that the
In the present study, to identify a wild ginseng-specific gene, we subtracted cDNAs expressed in wild ginsengs from those in cultivated ginsengs by using the SSH technique [15]. The technique of SSH is believed to generate an equalized representation of differentially expressed genes and to provide a high enrichment of differentially expressed mRNA. SSH overcomes the limitations of other gene analysis methods for differential expression. Its PCR-based approach allows for the effective removal of common genes from the RNA population prior to creating the library and has the advantage that reverse transcriptions are amplified efficiently [16].
We isolated a novel gene,
The