Fibrinolytic enzyme preparations were isolated from the snake venom of Macrovipera lebetica turanica in this study.
The purity of the preparations was determined using SDS-PAGE and the enzymic characteristics of the purified fibrinolytic enzyme were determined.
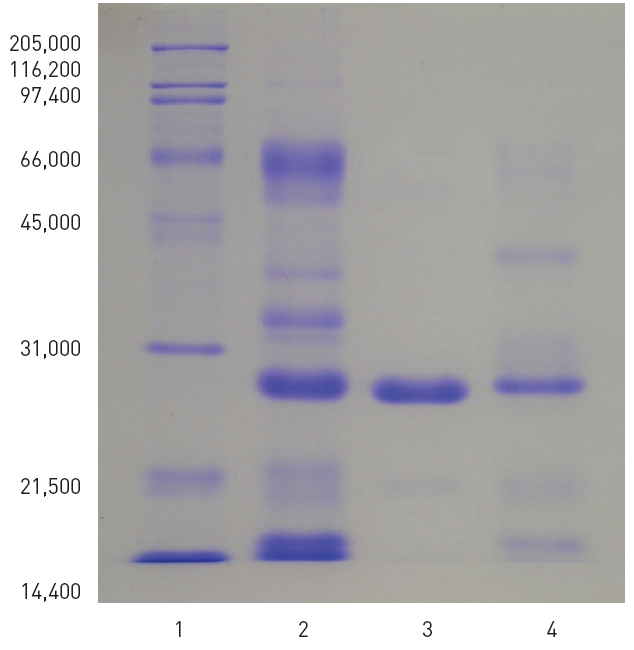
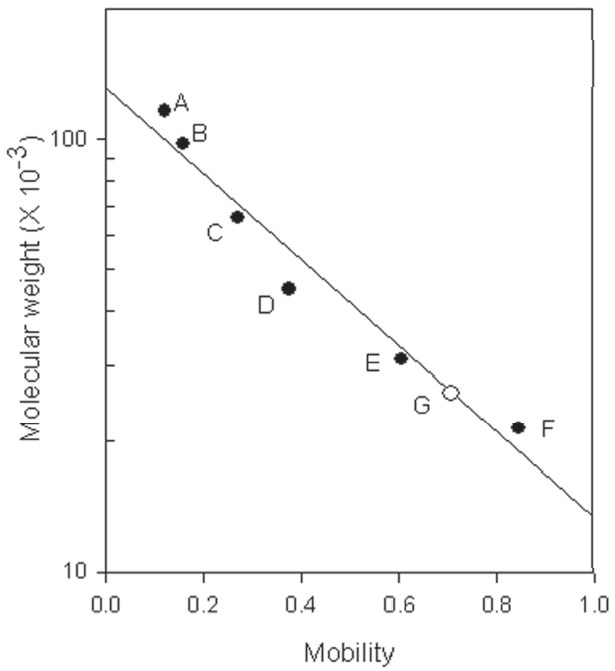
1. All of the two preparations with fibrinolytic activity obtained from the snake venom of M. l. turanicat contained the major polypeptide with the molecular weight of 27,500. One of the preparation showed purified fibrinolytic enzyme.
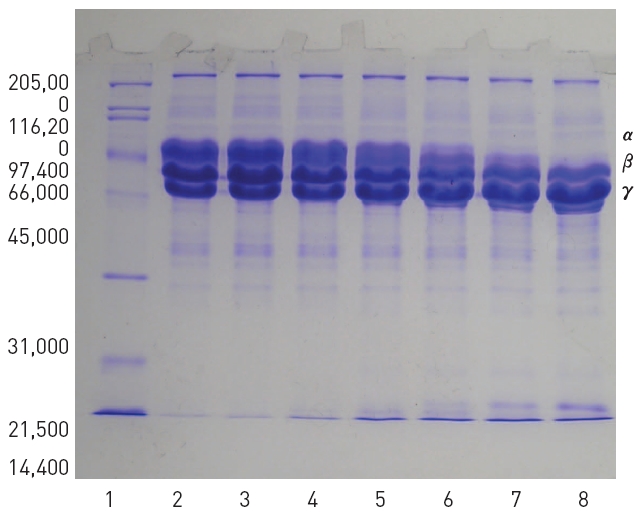
2. The purified fibrinolytic enzyme hydrolyzed α-chain of fibrinogen faster than β-chain but not γ-chain.
3. The fibrinolytic activity was inhibited completely by EDTA, EGTA, 1,10-phenanthroline, and dithiothreitol.
4. The fibrinolytic activity was inhibited completely by calcium chloride, iron(III) chloride, mercuric chloride, and cobalt (II) chloride.
5. The fibrinolysis zone formed after addition of zinc sulfate was smaller but clearer than the control.
These results suggested that the fibrinolytic enzyme purifed from the snake venom of M. l turanica was a metalloprotease containing dithiol group.
Snake venoms serve for the capture of small prey and are used defensively against large animals and humans. The neurotoxic venoms of cobras and related snakes (Elapidae) affect the nervous system, while the hemotoxic venoms of rattlesnakes and vipers (Viperidae) usually affect the circulation system 1). Snake venoms consist of a complex pool of proteins, organic compounds with low molecular weight, inorganic compounds.2),3) Proteins include enzymes, non-enzymatic proteins, enzymatic inhibitors, and peptides, many of which have found uses in the diagnosis and treatment of haemostatic disorders.4) Some of these toxins, both enzymic and non-enzymic, lead to the spectacular changes in haemostasis and the frequent hemorrhage seen after snake bite.5) It has been suggested that this process is part of a strategy to immobilize prey and to increase target tissue permeability to other venom toxins.6)The activities of these toxins include coagulation and anticoagulation of blood, platelet-activation, anti-platelet function, fibrinolytic activation, and hemorrhage.7)
The venom enzymes toxic to animals comprise acetylcholinesterases, ADPases, phopholipases, hialuronidases, and hemostatic proteases.2)The hemostatic proteases can be divided into metalloproteases and serine proteases, of which fibri(gen)olytic proteases have drawn particular attentions as potential therapeutic agents to remove plasma fibrinogen from the circulation for treatment of acute isochaemic stroke8) and to dissolve blood clot for treatment of occlusive thrombi.4)Fibrolase from copperhead snake venom degrades both α and β chains of fibrin and showed some promise as a thrombolytic agent.9) Alfimeprase, a recombinant fibrinolytic enzymes derived from fibrolase has been being developed as a clinical agent.10),11) Other fibrinolytic proteases which dissolve blood clots include afaacytin, from horned viper12) and the fibrinogenase from Macrovipera lebetina venom13).
The venom of
Lebetase, an α(β)-fibrin(ogen)olytic metalloprotease, possesses direct-acting fibrinolytic activity which involves direct cleavage of fibrin without plasminogen activation and is also an anticoagulant that acts by inhibiting platelet aggregation. The enzyme readily hydrolyzes the Aα chain and more slowly the Bβ-chain of fibrinogen and the oxidized insulin B-chain in the positions Ala14-Leu15 and Tyr16-Leu17. Its fibrinolytic activity is inhibited by EDTA and dithiothreitol. The protease consists of a single polypeptide chain with a molecular weight of 23,700 and is a metalloenzyme that contains one mole of Zn2+ and one mole of Ca2+ per mole of protein.17),18)The mature protein lebetase consists of 204 amino acids and its amino acid sequence shows strong similarity with fibrolase. The metallo protease domain has a typical zinc-chelating sequence.19)
The present study isolated a fibrinolytic enzyme from
The snake venom from
2. Purification of fibrinolytic enzyme
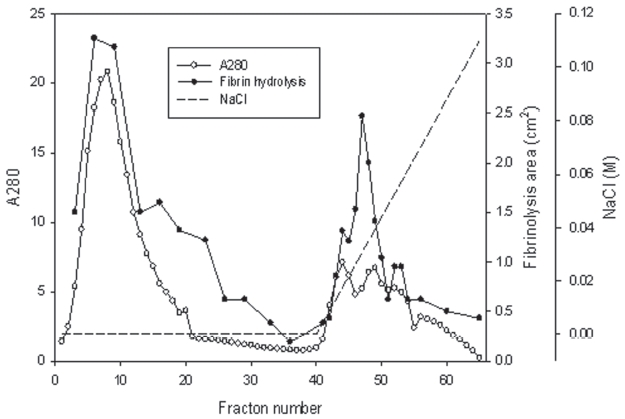
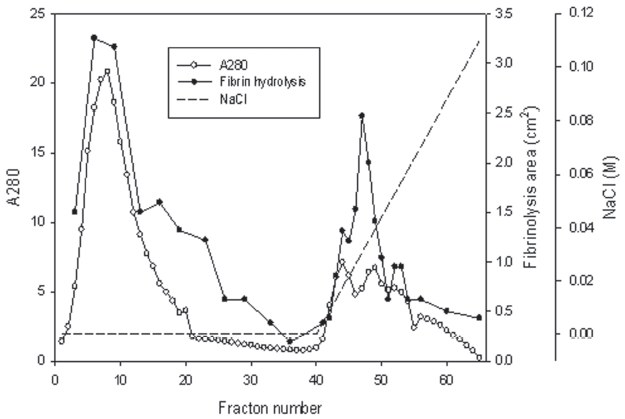
The venom powder (2 mg) was dissolved and dialyzed in 50 mM Tris-HCl, pH 7.6. The venom solution was loaded on the column (5 × 5.8 cm) of Q-Sepharose (GE, USA) equilibrated with the buffer. The column was eluted with the buffer and then with a concentration gradient of sodium chloride of up to 0.3 M. Two fractions with fibrinolytic activity were obtained (Fig 1).
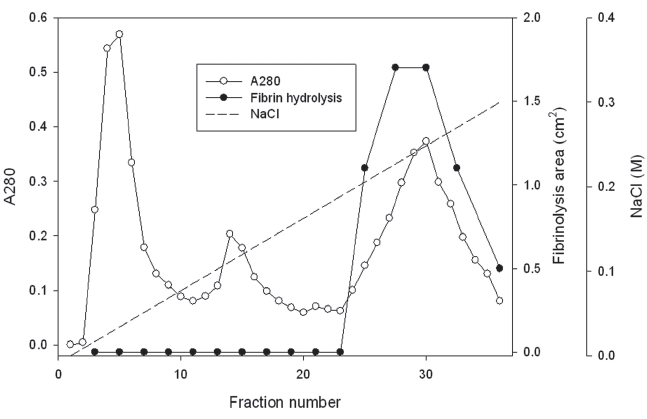
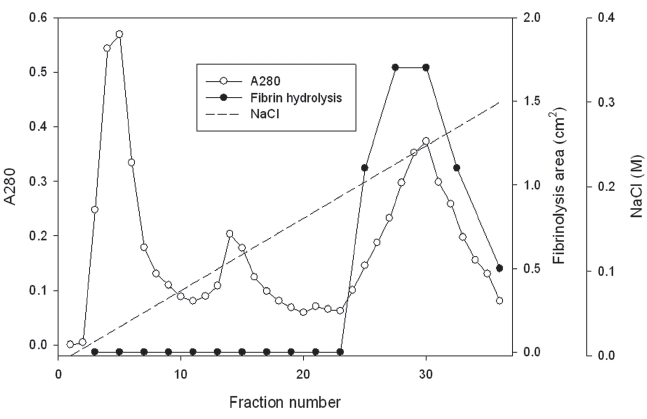
The first fraction eluted with 50 mM Tris-HCl, pH 7.6 was dialyzed in 50 mM Tris-HCl, pH 8.6. The dialyzed solution was loaded on a column (2.5 × 10 cm) of Q-Sepharose equilibrated 50 mM Tris-HCl, pH 8.6. The column was washed with the buffer and eluted with a concentration gradient of sodium chloride up to 0.2 M (Fig 2). The fraction with fibrinolytic activity was dialyzed in 50 mM Tris-HCl, pH 7.6, 0.15 M NaCl and separated on the column (2.5 × 120 cm) of Sephadex G-75.
The second fraction released with the concentration gradient of sodium chloride from the column of Q-Sepharose (Fig 1) was dialyzed in 50 mM Tris-HCl, pH 7.6, 0.15 M NaCl and were separated on the column (2.5 ×120 cm) of Sephadex G-75 (GE, USA) equilibrated with the buffer. All the chromatography was performed at the refrigeration temperature.
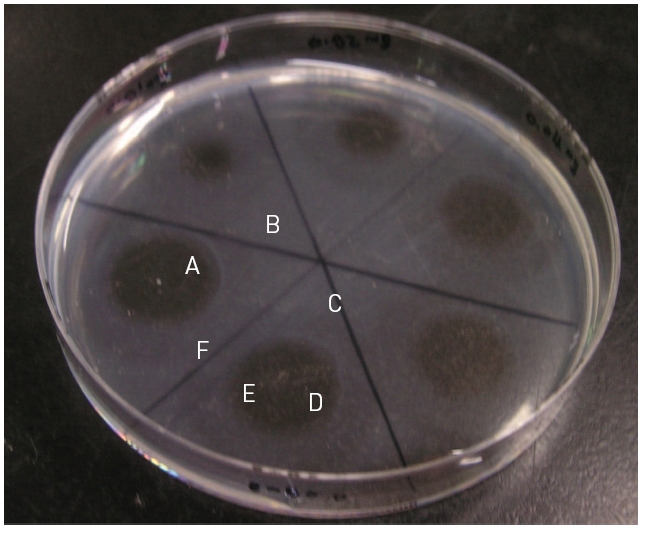
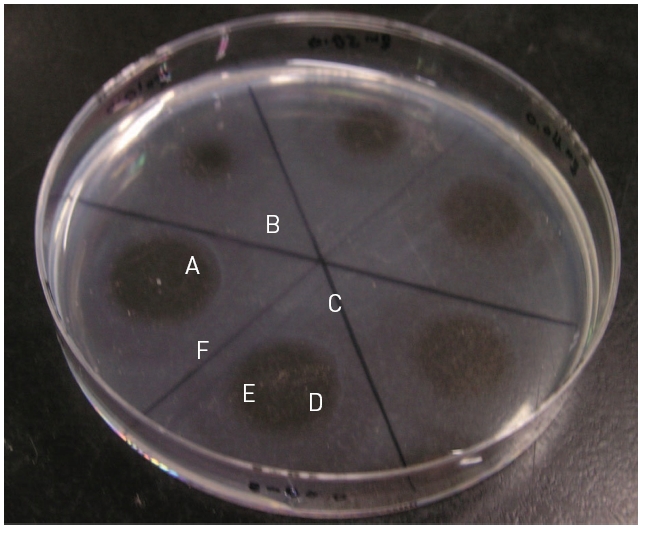
Fibrinolytic activity was determined on fibrin plates according to the procedure of Astrup and Mullertz.21) An aliquot (200 μl) of thrombin (Sigma-Aldrich, USA) solution (10 U/ml in 100 mM Tris-HCl, pH 7.8) were added to 9 ml of 0.1% fibrinogen (Sigma-Aldrich, USA) dissolved in the buffer solution. The mixed solution was added into petri dish with the diameter of 9 cm and incubated at 37°C for 1 hr, until it converted into fibrin gel. An aliquot (10 μl) of the venom fraction was placed on the fibrin gel and then incubated at 37°C for 18 hr. Diameters (r1 and r2) at a right angle of hydrolysis zone were measured. The fibrinolysis area was calculated following the formula of 0.785 r1 × r2.
Sodium dodecyl sulfate - polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to the procedure described by Laemmli.22)The molecular weight markers (Bio-Rad, USA) for the SDS-PAGE consisted of myosin (200,000), β-galactosidase (116,250), phosphorylase b (97,400) bovine serum albumin (66,200), ovalbumin (45,000), carbonic anhydrase (31,000), trypsin inhibitor (21,500), lysozyme (14,000), and aprotinin (6,500).
The fibrinogen (4 mg) in 200 μl of 50 mM Tris-HCl, pH 7.5, 0.15 M NaCl was added with 50 μl of the purified fibrinolytic enzyme and then the mixture was incubated at 37°C for 6 hr. An aliquot (10 μl) of the mixture was taken at 0, 10, 30, 60, 120, and 240 min during the incubation and added with equal amount of the sample buffers for SDS-PAGE each time. The mixtures were added with a couple of drops of mineral oil, heated at 100°C for 5 min, and analyzed in SDS-PAGE.
Protein concentration was determined using BCA Protein Assay Reagent (Pierce, USA). An aliquot (100 μl) of samples was mixed with 2.0 ml of BCA Reagent which is prepared following the producer’s manure. The mixture was incubated at 37°C for 30 min and then cooled at room termperature for 30 min. The absorbance at 562nm of the mixture was measured. Bovine serum albumin was used for calibration curve.
1. Purification of fibrinolytic enzyme from the snake venom
The snake venom was separated into two major fractions by using ion exchange chromatography of Q-Sepharose which was eluted using 50 mM Tris-HCl, pH 7.6 and then using the buffer with a concentration gradient up to 0.1 M sodium chloride. The first fraction from 4 to 23 and the second fraction from 44 to 53 showed strong fibrinolytic activity in fibrin plate assay (Fig 1).
The first fraction was dialyzed in 50mM Tri-HCl, pH 8.6. The dialyzed sample was separated on a column of Q-Sepharose equilibrated with the buffer (Fig 2). The fraction release from the column with the buffer did not show fibinolytic activity (results not shown). The column was then eluted using the buffer with a concentration gradient of sodium chloride up to 0.3 M sodium chloride. The chromatogram in Fig 2 showed three fractions released from the column. The last fraction from 25 to 36 showed fibrinolytic activity in fibrin plate assay.
The second fraction in Fig 1 and the last protein fraction in Fig 2 were were dialyzed in 50 mM Tris-HCl, pH 7.6, 1.5 M NaCl, and then subjected to gel filtration chromatography using the column of Sephadex G-75, respectively (results not shown). The fractions with fibrinolytic activity were concentrated in dialysis bag using polyethylene glycol, and frozen stored.
The volume and protein concentration of the fibrinolytic enzyme preparations isolated originally from the first fraction and the second fraction of ion exchange chromatography of Q-Sepharose in Fig 1 were 6.7ml and 3.29 mg/ml and 15.4ml and 3.92 mg/ml, respectively.
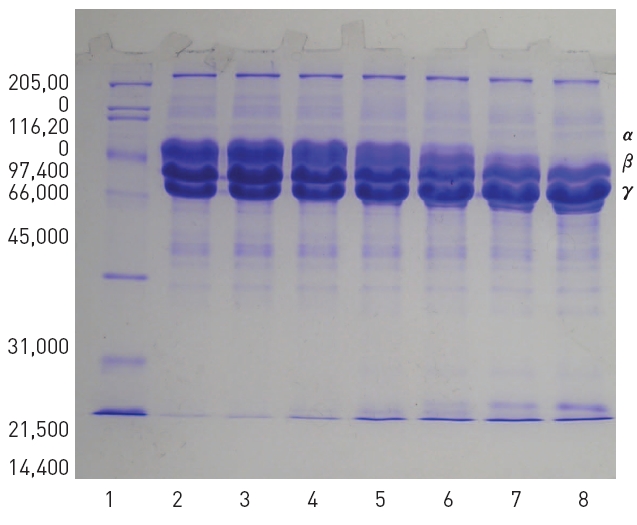
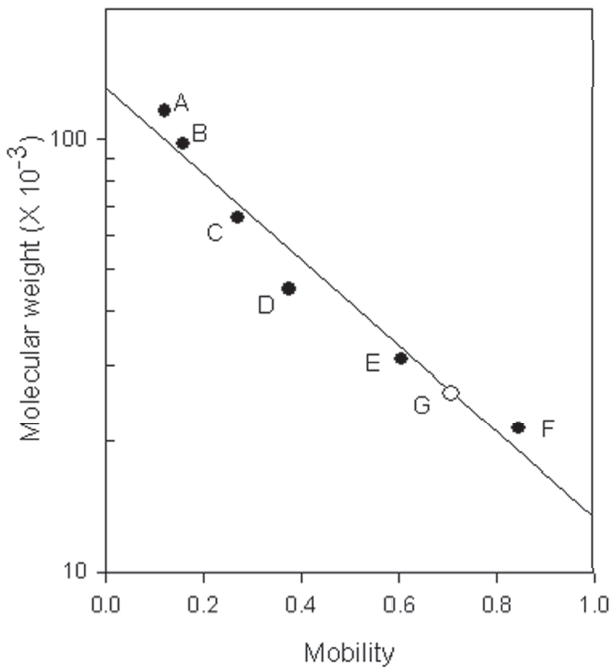
SDS-PAGE of venom of
Two hundred μl of 2% fibrinogen was added with 50 μl of the purified fibrinolytic enzyme and then the mixture was incubated at 37 °C for 4 hr. Ten μl of the mixture was taken at 0, 10, 30, 60, 120, and 240 min during the incubation and subjected to SDS-PAGE. The SDS-PAGE in Fig 5 showed that α-chain of fibrinogen was hydrolyzed preferentially and no intact α-chain was left after incubation for 240 min. β-Chain of fibrinogen was hydrolyzed more slowly than α-chain but γ-chain of fibrinogen was not hydrolyzed.
4. Inhibition of the fibrinolysis by protease inhibitors and salts
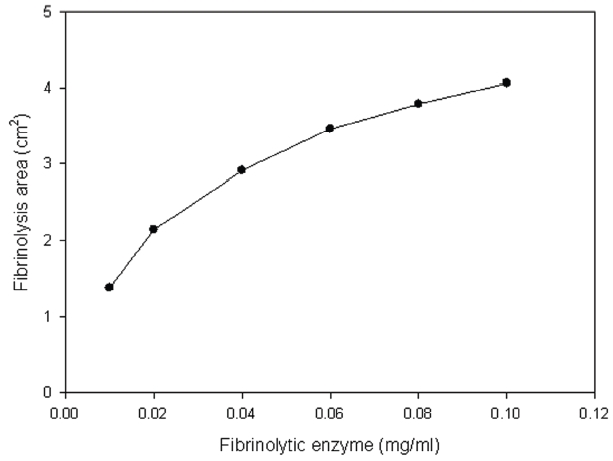
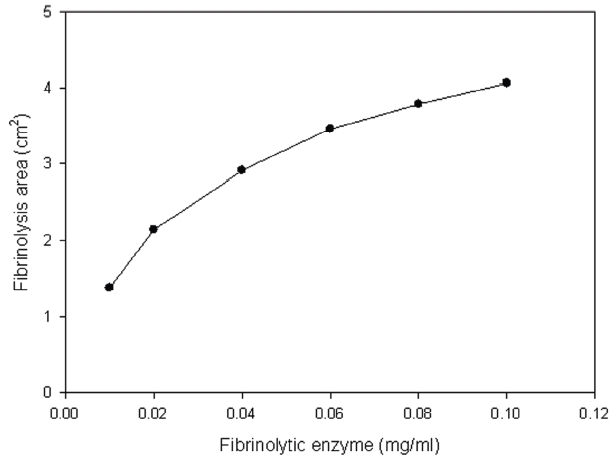
Thrombin was added into fibrinogen solution and then the mixture was incubated at 37°C for 1 hr and then fibrinogen formed a translucent fibrin gel in the fibrin plate assay (Fig 6). The fibrinolytic enzyme which was placed on the gel hydrolyzed the fibrin gel and made a transparent circular zone filled with liquid as shown in Fig .6The relationship between the area of the zone which was calculated from the measured diameter and the concentration of the fibrinolytic enzyme was shown in Fig .7The results showed that the fibrinolysis area increased steeply at the lower concentrations from 0.01 to 0.02 mg/ml and then the slope became lower at the higher concentrations.
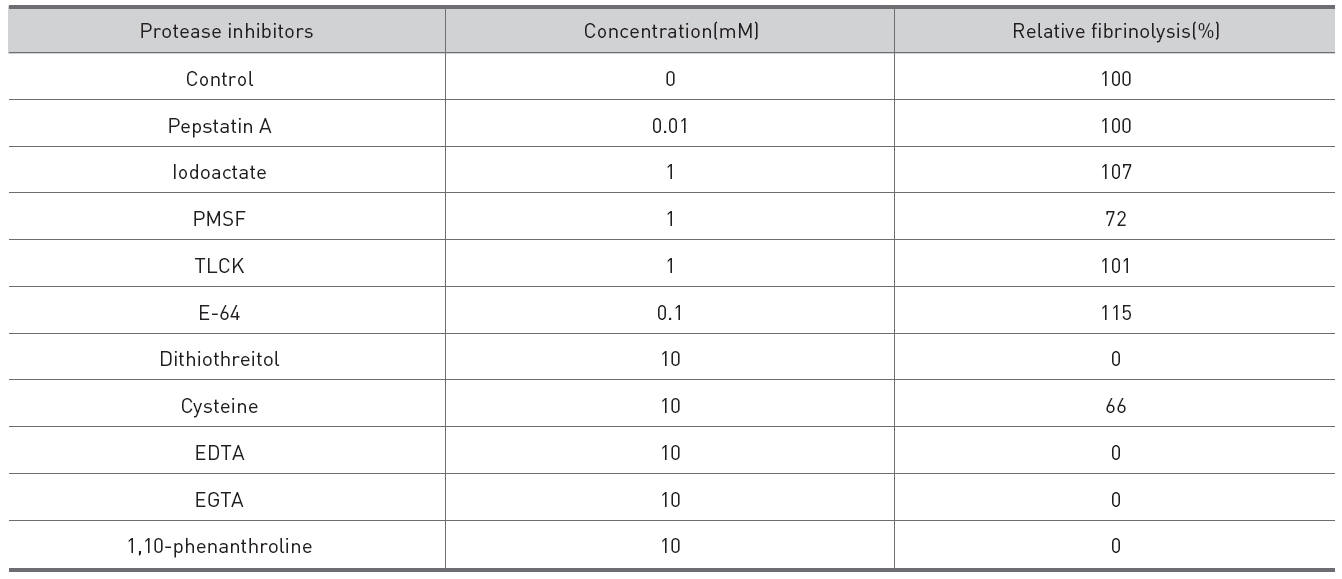
Protease inhibitors were added to the fibrinolytic enzyme of 0.02 mg/ml and 10μl of the mixed solutions were placed on the gel in fibrin plate assay to determine their inhibitory effects on the fibrinolysis (Table 1.). The metalloprotease inhibitors, EDTA, EGTA, and 1,10-phenanthroline, inhibited the fibrinolysis completely. The dithiol-reducing compounds, cysteine and dithiothreitol, showed partial and complete inhibition on the fibrinolysis, respectively. Serine protease inhibitors, PMSF and TLCK, an acid protease inhibitor, pepstatin A, and a cysteine protease inhibitor, E-64, showed few inhibition on the fibrinolysis.
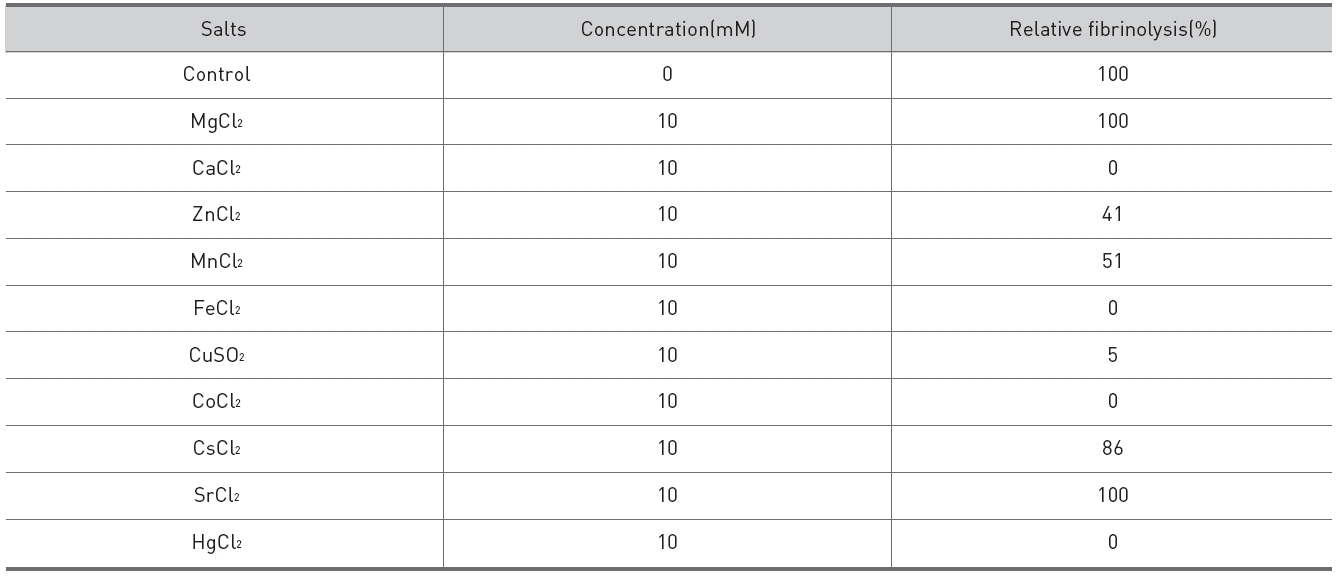
Salts of 10 mM were added to the fibrinolytic enzyme of 0.02 mg/ml and 10 μl of the mixed solutions were placed on the gel in fibrin plate assay to determine their inhibitory effects on the fibrinolysis. Calcium chloride, iron(III) chloride, mercuric chloride, and cobalt (II) chloride inhibited the finolysis completely. Cupric sulfate inhibited the fibrinolysis strongly. Manganese chloride, zinc sulfate, and cesium chloride showed partial inhibition on the fibrinolysis. Strontium chloride and magnesium chloride did not inhibit the fibrinolysis. The fibrinolysis zone which was formed after addition of zinc chloride was clearer than the control.
The venom of
Lebetase, is an α(β)-fibrin(ogen)olytic metalloprotease, possesses direct-acting fibrinolytic activity which involves direct cleavage of fibrin and hydrolyze α-chain of fibrinogen faster than β-chain of fibrinogen. Its fibrinolytic activity is inhibited by EDTA and dithiothreitol. The protease consists of a single polypeptide chain with a molecular weight of 23,700 and is a metalloenzyme that contains one mole of Zn2+ and one mole of Ca2+ per mole of protein.18),19)
Two major fractions with fibrinolytic activity were separated from the venom of
The fibrinolytic enzyme purified from the venom of
Its fibrinolytic acitivity was inhibited completely by calcium chloride completely. The hydrolysis zone formed after addition of zinc chloride was smaller but clearer than the control. These results suggested that the fibrinolytic enzyme preparation might have lost some cationic salts during the purification, if the fibrinolytic enzyme were lebetase. Further studies are necessary to determine the effects of salts on the fibrinolysis.
In the present study a fibrinolytic enzyme was purified from the venom of
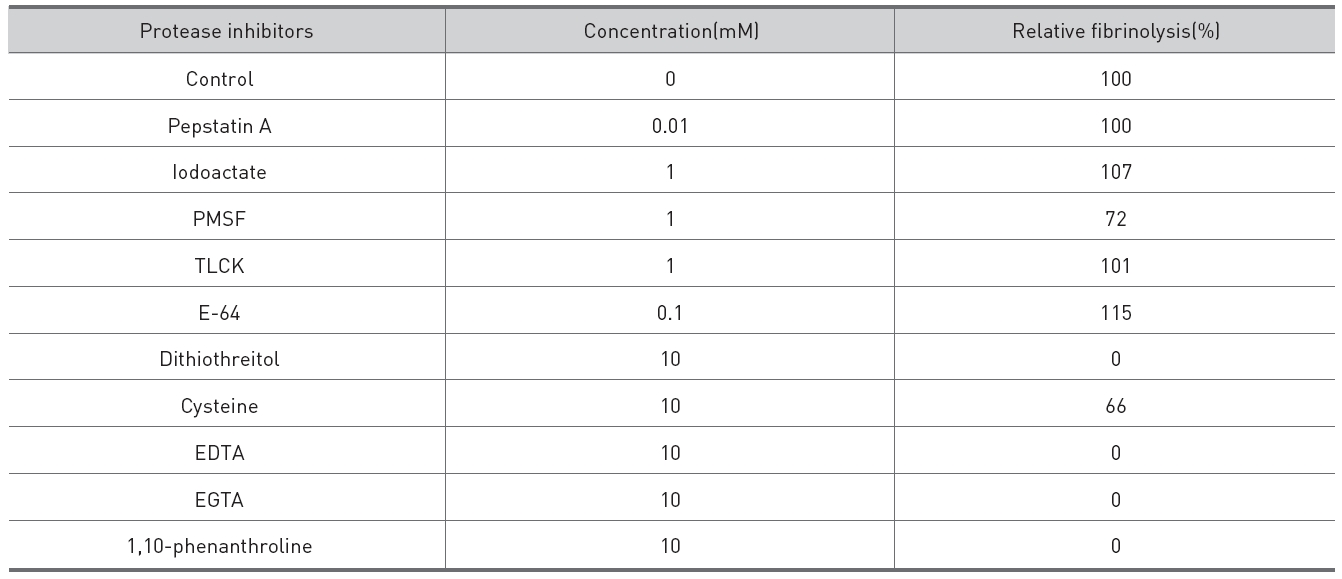
Effects of protease inhibitors on the fibrinolysis by the fibrinolytic enzyme purified from the venom of M. l. turanica.
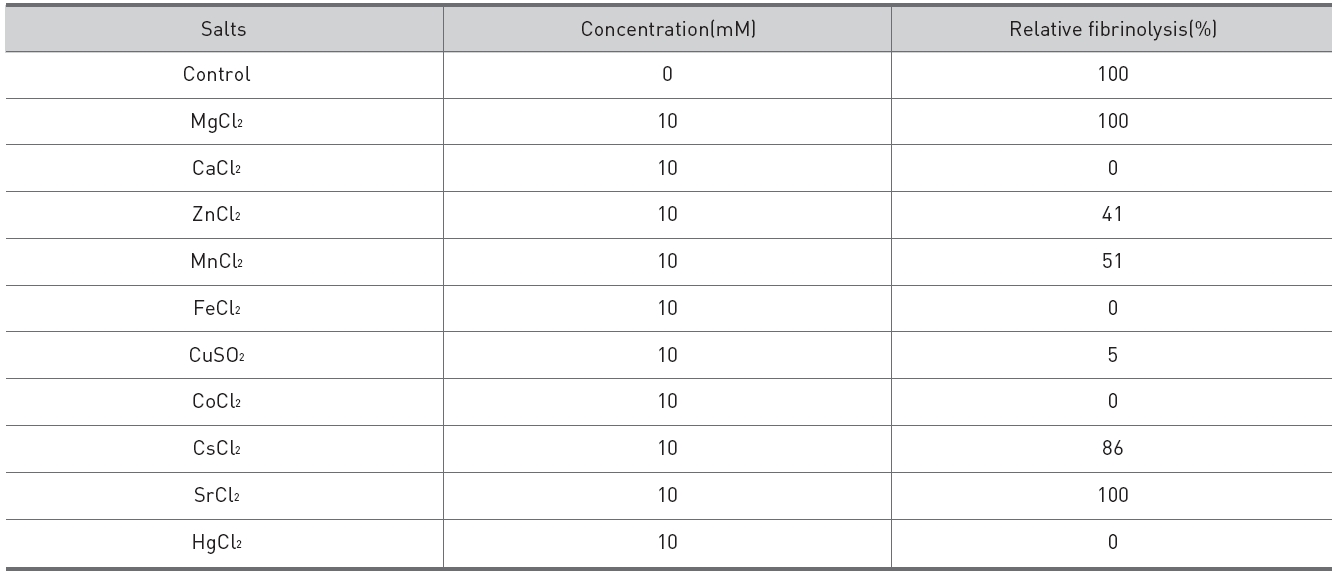
Effects of salts on the fibrinolysis by the fibrinolytic enzyme purified from snake venom of M. l. turanica