Marine microalgae are a key diet component in finfish and shellfish aquaculture. Cryopreservation of the microalgae is suggested by many other studies as the best method for long-term storage. To test cryopreservation efficacy, 19 taxas of marine microalgal species were examined. In the first experiment we compared dimethylsulfoxide (Me2SO) and glycerol, which are most widely used as cryoprotectant agents (CPAs). The cryopreservation comprised two freezing procedures. Firstly, the samples containing the CPAs were kept at 4℃ for 10 min before being plunged into liquid nitrogen (-196℃). Secondly, samples containing CPAs were pre-cooled (-1℃ min-1) to -80℃ before being plunged into liquid nitrogen. Most of the species were successfully cryopreserved using Me2SO, whereas the Prasinophyceae (T. striata and T. suecica) were successfully cryopreserved using glycerol. In general, the cooling method had no influence on the survival of the microalgae except in the case of the Tetraselmis species. In the second experiment, the cultured solution was divided before cryopreservation into concentrated and non-concentrated groups to identify the effect of cell density during cryopreservation. After 12 months of storage, the samples were again divided into centrifugation and non-centrifugation groups to learn the effect of Me2SO on the culture. Viability and growth of the microalgae were not influenced by cell density or the centrifugal removal of the Me2SO after thawing.
Marine microalgal species are widely used as a foodsource in the artificial propagation of hatchery-rearedmollusks, crustaceans and fish (Canavate and Lubian1995b) and could be the source of diverse new productsand medicines (Rhodes et al. 2006). For this reason, specialcontrol of microalgal species is needed to ensurelong-term storage free of any contaminants.
One preservation method for microalgae is to use a liquidor agar medium. Compared to freezing (0°C to -20°C), which is effective for only about two months(Terauchi et al. 1997), storage is possible for up to a yearwhen an agar medium is used. But long-term storage ofsubcultures runs the risk of contamination from differentorganisms and genetic mutation (Nagasaki 2001).Therefore, a long-term preserving method for microalgaeis important. Recently in aquaculture, drying, freeze-drying,and cryopreservation techniques have beenexplored. However, drying and freeze-drying have notbeen very successful for the long-term storage of algae(Taylor and Fletcher 1998).
Cryopreservation has been successfully used in animalsand plants (Taylor and Fletcher 1998). Reports onthe cryopreservation of microalgae have recentlyincreased (Poncet and Veron 2003; Houdan et al. 2005).Cryopreservation entails preparing the cultures and thenstoring them in liquid or vapor-phase nitrogen. Thesemethods require significant initial effort, but in the longtermreduce labor requirements and opportunities forcontamination. In addition, because these are long-termstorage methods, genetic diversity is maintained.
A few freshwater microalgal species and some marinediatoms have been successfully cryopreserved, but it iswidely recognized that many microalgal species arerecalcitrant to standard cryopreservation techniques(Canavate and Lubian 1995a, 1995b, 1997a, 1997b;Tzovenis et al. 2004). The importance for the survival ofthe microalgae of the interaction between the rate ofcooling, the cryoprotective additive and the rate ofwarming has been stressed (Mazur 1969).
This study describes differences in the viability and growth of marine microalgae when subjected to different cryoprotectant agents (CPAs), cooling methods, cell densities at the time of cryopreservation, and the removal of the dimethylsulfoxide (Me2SO) after thawing.
>
Organisms and culture conditions
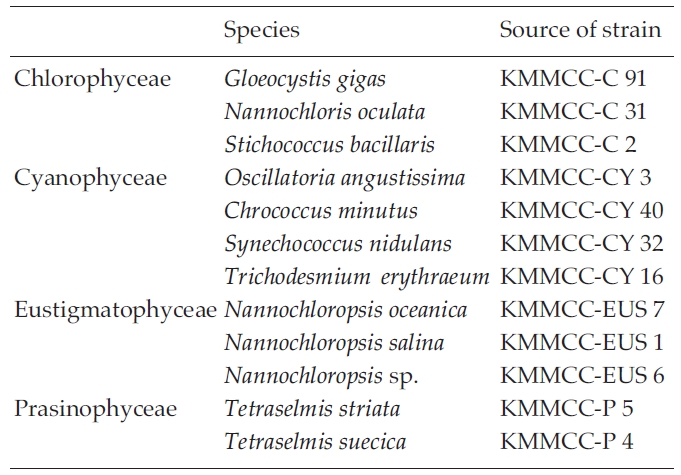
In the first experiment on cryopreservation freezing procedures, 12 marine microalgal species were examined (Table 1): Gloeocystis gigas, Nannochloris oculata, Stichococcus bacillaris, Nannochloropsis oceanica, N. salina, N. sp., Chrococcus minutus, Oscillatoria angustissima, Synechococcus nidulans, Trichodismium erythraeum, Tetraselmis striata and T. suecica. All species were maintained at the Korea Marine Microalgae Culture Center (KMMCC). The microalgae were grown in 250 mL Erlenmeyer flasks containing 50 mL of f/2 medium filtered seawater (33 psu, Guillard and Ryther 1962) at 20℃ with continuous illumination of 60 μmol photon m-2 s-1 provided by white fluorescent light without aeration for 7 days.
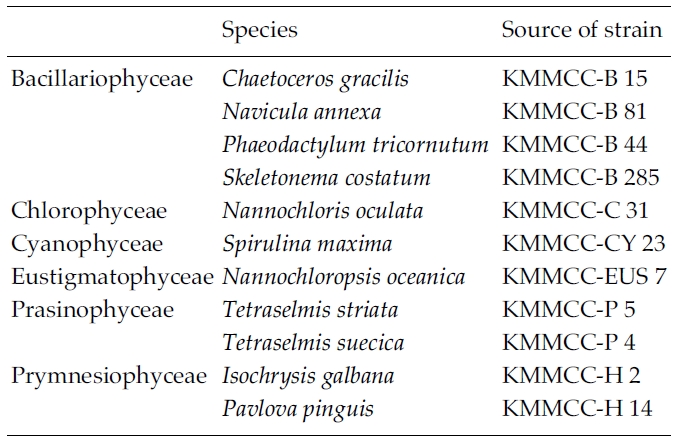
To test the effect of cell density during cryopreservationand the removal of Me2SO after thawing on the viabilityand growth of the microalgae, 11 microalgalspecies at the KMMCC were examined (Table 2):Chaetoceros gracilis, Navicula annexa, Phaeodactylum tricornutum,Skeletonema costatum, N. oculata, Spirulina maxima,N. oceanica, T. striata, T. suecica, Isochrysis galbana, andPavlova pinguis. The culture condition of the microalgaewas the same as those mentioned-above except for S.maxima, which was cultivated in Spirulina medium(Schlosser 1982). To test the effect of cell density duringcyropreservation, two samples were used as a controlgroup: one in which the cell density of the culture wasconcentrated three times, the other in which the culturewas non-concentrated. For this test, liquid cultures wereharvested at exponential growth phase by centrifucation(1,000 rpm, 5 min for Spirulina and 1,500 rpm, 10 min forothers species). The concentrated microalgae werewashed with the fresh medium three times and the pelletswere resuspended in fresh medium.
In the first experiment, we used two different cryoprotectant agents: dimethylsulfoxide (Me2SO, 10%) and glycerol (10%). Filtered Me2SO was added to the autoclaved f/2 medium. Glycerol was added to the f/2 medium, which was then autoclaved. Samples of the culture (0.5 mL) were dispensed into cryovials (T310-2A 2 mL,
Micoralgal species used in the experiment on the effectof cryoprotective agents and freezing procedures
Simport, Canada) and incubated at 4°C for 10 min priorto cryopreservation. Cryopreservation consisted of twofreezing methods: one, the samples containing the CPAswere transferred and held at 4°C for 10 min before beingplunged into liquid nitrogen (-196°C); two, the samplescontaining CPAs were transferred to hold at 4, -20 and -80°C for 10 min, respectively before being plunged intoliquid nitrogen. Their viability and growth were testedafter 1 and 6 months of storage.
In the second experiment, 10% Me2SO was used for the CPA. The samples containing the Me2SO was kept at 4℃ for 10 min before being plunged into liquid nitrogen (-196℃). Their growth was tested after 12 months of storage in liquid nitrogen.
After one and six months of storage, samples of the frozen paste were extracted from the liquid nitrogen and transferred immediately to a water bath at 35℃ for 5 min. To test the viability, cells were thawed at 35℃ until the ice was completely melted. Thawed samples were diluted ten-fold in fresh f/2 medium. The samples were incubated at 20℃ with an illumination of 60 μmol photon m-2 s-1. The cells were counted on a Neuvauer haemacytometer for 15 days and then we analyzed the daily specific growth rate (s.g.r = 3.322 x log(N1/N0)/t1- t0) (Guillard 1973) during log phase growth. Motile Prasinophyceae (T. striata and T. suecica) were treated with 1% formaldehyde and the total number of cells was counted. All the experiments were performed in triplicate.
After 12 months of storage, the cells were thawed by
Micoralgal species used in the experiment on the effectof cell density in cryopreservation and removal of Me2SOafter thawing
the same method mentioned-above. The samples weredivided into two groups to learn the effect of the Me2SOon the culture. The thawed samples were directly dilutedin fresh f/2 or Spirulina medium. The other sampleswere harvested by centrifugation (5,000 rpm, 3 min) andwashed once with fresh medium to remove the Me2SO.The pellets were resuspended in fresh medium and cultured.The cells were cultured by using the previousmethod. The cells were counted every 5 days for 60 daysand daily s.g.r was analyzed during log phase growth.Motile Prasinophyceae (T. striata, T. suecica) andPrymnesiophyceae (I. galbana, P. pinguis) were treatedwith 1% formaldehyde and the total number of cells wascounted. All the experiments were performed in triplicate.
The statistical significance level was analyzed by onewayanalysis of variance (ANOVA) (Duncan’s multiplerange tests; Duncan 1955). Statistical significance wasdetermined at p < 0.05. Statistical analysis of data includedthe Student’s t-test. All statistics were conducted bySPSS software (version 10.1; SPSS Inc., Chicago, IL,USA).
>
Effect of CPAs and cooling methods Growth after cryopreservation for one month
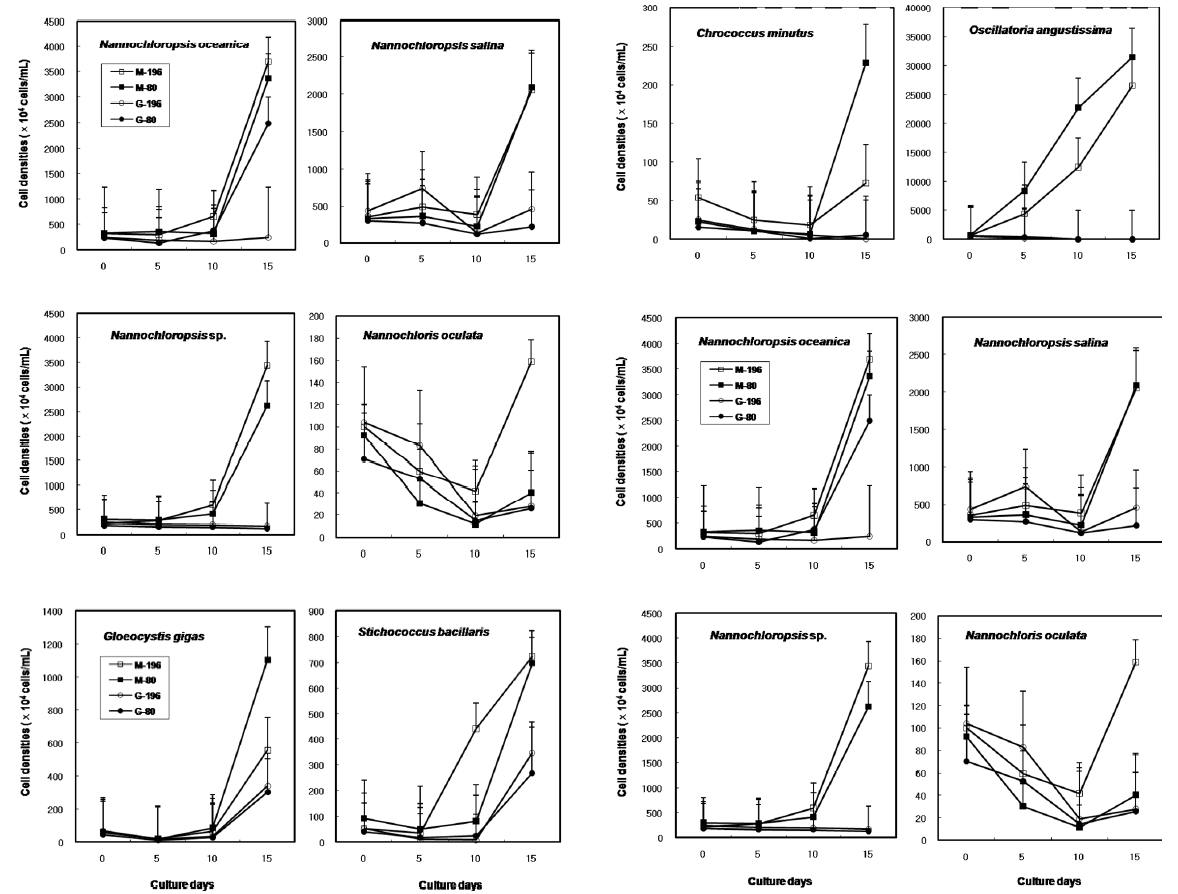
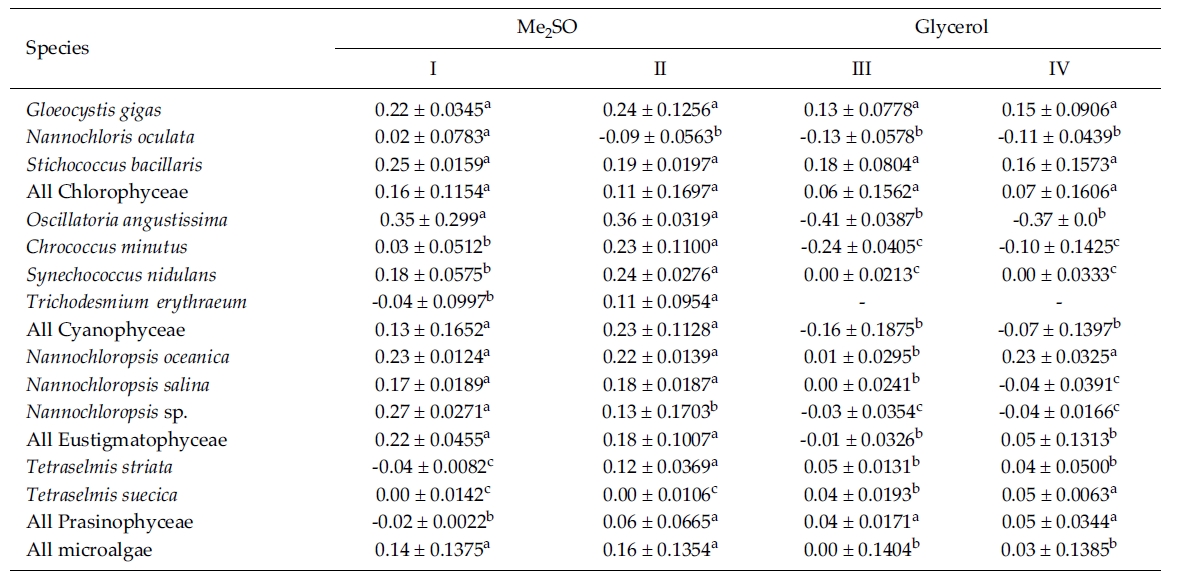
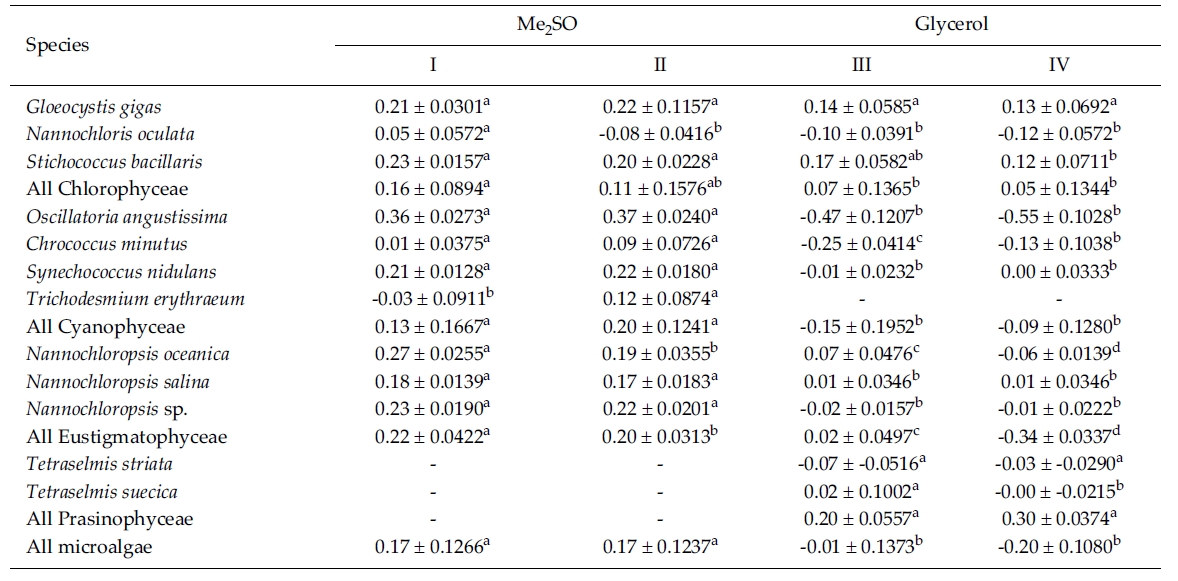
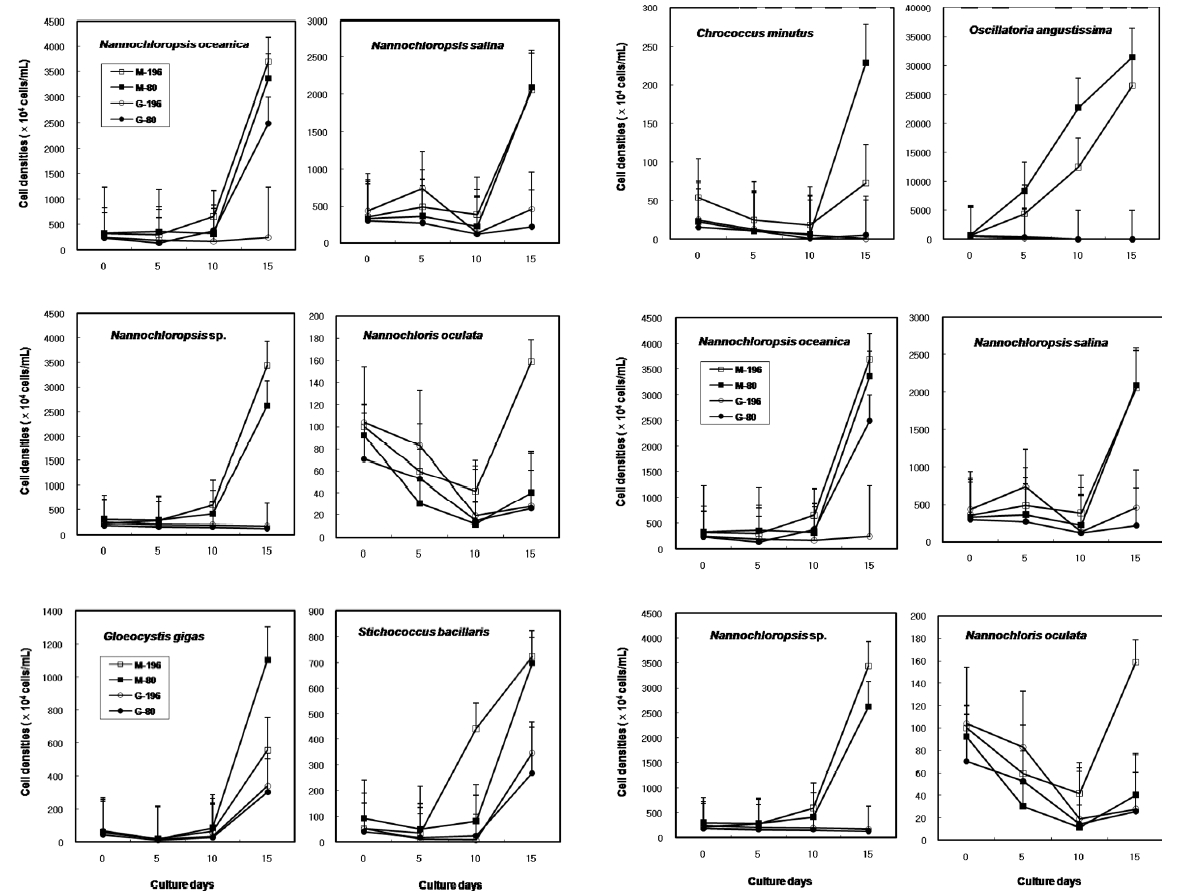
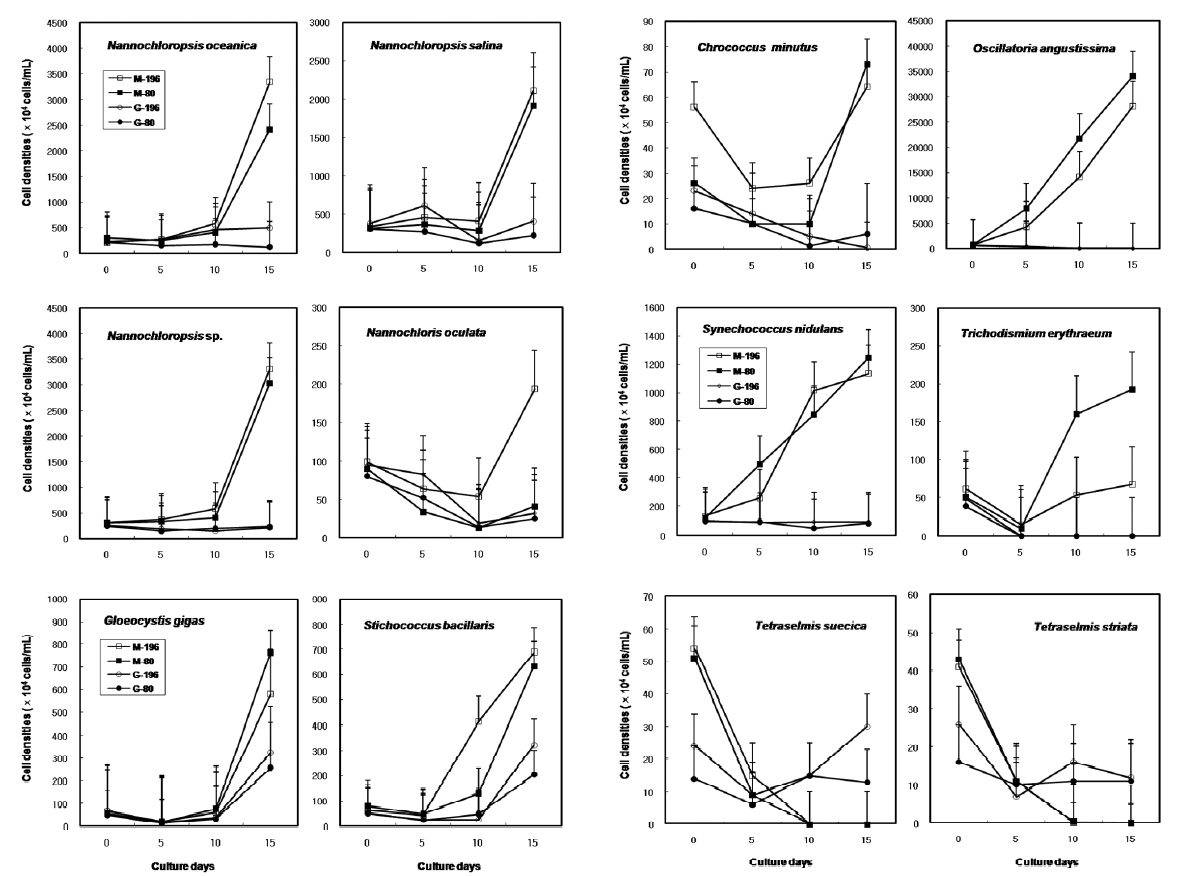
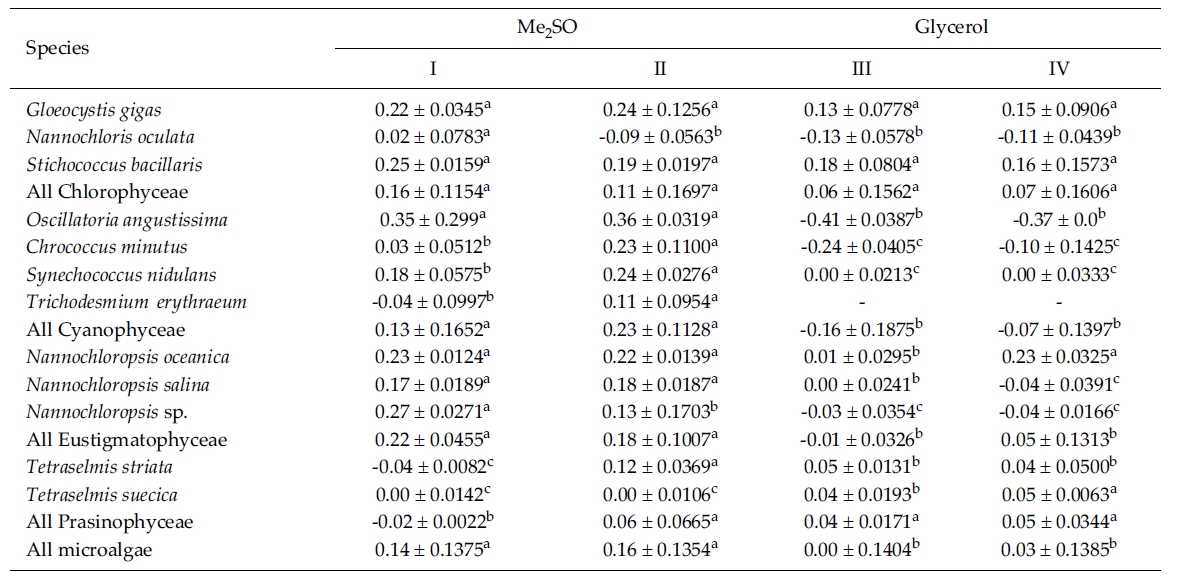
Most of the species were successfully cryopreserved using 10% Me2SO (Fig. 1). Growth of N. oceanica proved best following a rapid cooling from 4°C and then a plunge into liquid nitrogen with the addition of Me2SO. Its cell density reached to 3433.3 x 104 cells mL-1. However, when using glycerol, growth differed depending on the freezing procedure and microalgae growth was very low with 129.1 x 104 cells mL-1 when the rapid cooling method was used. The effects of freezing procedures on the growth of cryopreserved N. salina were not different in Me2SO. The effects of slow and rapid cooling on the growth of cryopreserved N. sp. were different in Me2SO. But no growth was found in glycerol. N. oculata grew only following rapid cooling in Me2SO. G. gigas grew best in Me2SO with slow cooling. The cooling procedure did not affect the growth of G. gigas in glycerol. The effects of slow and rapid cooling on the growth of cryopreserved S. bacillaris were not different in any of the CPAs. But Me2SO showed higher growth than glycerol did.
C. minutus grew to 228.5 x 104 cells mL-1 in slow coolingwith Me2SO. O. angustissima was successfully cryopreservedin Me2SO, but the effect of cooling method wasnot distinct. S. nidulans and T. erythraeum did not grow inglycerol. The effects of slow and rapid cooling on thegrowth of these microalgal species were different whentried in Me2SO. The cell density of S. nidulans in slowcooling with Me2SO reached to 1365.8 x 104 cells mL-1.Although the growth was low in Tetraselmis, the additionof glycerol in rapid cooling method increased the celldensity of T. suecica to 39.0 x 104 cells mL-1.
The daily specific growth rate of each microalgal group by class in this experiment is shown in Table 3. In general, Me2SO produced significantly higher growth rate than glycerol (p < 0.05); however, the cooling method had no affect on the growth of the microalgae. In Eustigmatophyceae and Cyanophyceae, cryopreservation with Me2SO showed significantly higher growth rate than that with glycerol (p < 0.05). In Chlorophyceae, the choice of the CPA and cooling method had no affect on the growth of the microalgae. Cryopreservation of Prasinophyceae was more difficult than that for the other microalgal groups.
>
Growth after six-month of cryopreservation
The results on growth after cryopreservation for six month is shown in Fig. 2. Although N. oceanica grew well to the density of 3340 x 104 cells mL-1 in Me2SO following the rapid cooling method, it grew badly in glycerol and the freezing methods were not distinct. The effects of slow and rapid cooling on the growth of cryopreserved N. salina and N. sp. were not distinct in Me2SO. But glyc-erol showed bad growth. N. oculata grew best in Me2SOfollowing the rapid cooling method. G. gigas was successfullycryopreserved using all cryoprotectant agentsand freezing procedures. The effects of slow and rapidcooling on the growth of cryopreserved S. bacillaris werenot distinct in any of the CPAs. But, glycerol produced alower growth than Me2SO.
The effects of slow and rapid cooling on the growth of C. minutus and S. nidulans were not distinct in any of the CPAs. Maximum growth of O. angustissima was recorded with 33980.0 x 104 cells mL-1 in Me2SO following slow cooling. T. erythraeum grew best in Me2SO using the slow cooling method. Tetraselmis grew better in glycerol than in Me2SO, but growth was very low less than 30.0 x 104 cells mL-1.
The daily specific growth rate of each microalgal group by class in this experiment is shown in Table 4. The effects of slow and rapid cooling on the growth rate of all the cryopreserved Chlorophyceae and Cyanophyceae were not significantly different for Me2SO and glycerol (p < 0.05). The growth rate of all the Eustigmatophyceae was the highest significantly in Me2SO following the rapid cooling method (p < 0.05). Prasinophyceae grew only in glycerol, but freezing method did not affect the growth rate. In general, the effects of slow and rapid cooling on the growth rate of most cryopreserved marine microalgae were not different to each other and in all cases and Me2SO proved a more effective growing medium than glycerol except Prasinophyceae.
>
Effect of cell concentrations in cryopreservation and removal of Me2SO after thawing
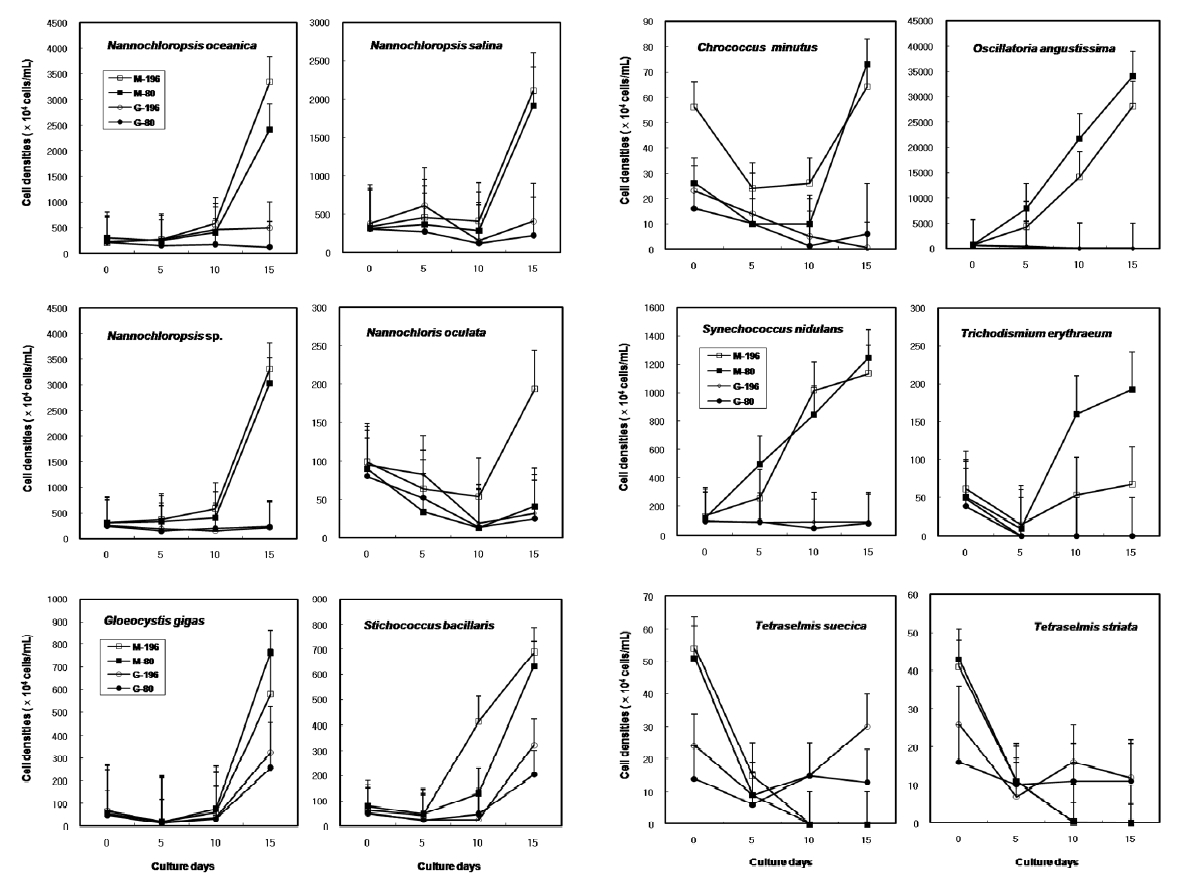
In this experiment we used 10% Me2SO as a CPA because most of the microalgae in the previous experi-ments were successfully cryopreserved using 10% Me2SO. The result of the specific growth rate of 11 microalgal species is shown in Table 5. The growth rate of C. gracilis with the Me2SO removed in a non-concentrated sample was significantly higher than that in a concentrated one (p < 0.05). N. annexa showed the highest
Mean daily specific growth rates for fifteen-day cultures of each microalgal species cryopreserved with different cryoprotectantagents and cooling methods for one month (I and III, kept in -196°C directly; II and IV, kept in -196°C after pre-cooling(-1°C min-1) to -80°C. No living cell was found on the final culture day. A different superscripted lower case letter in the samerow means a significant difference at the p < 0.05 level)
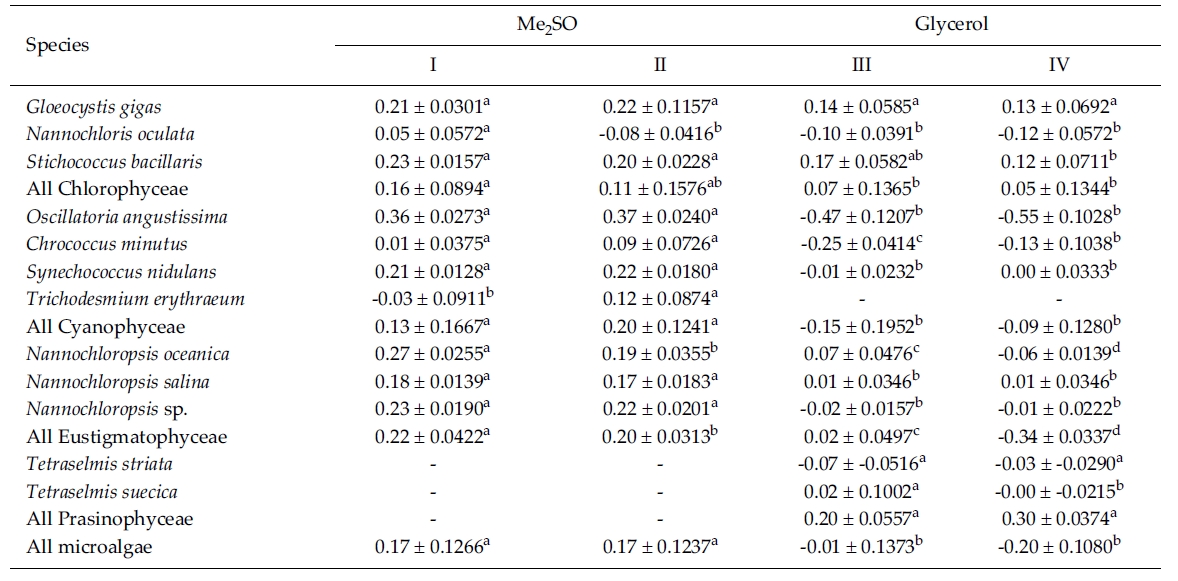
Mean daily specific growth rate for fifteen-day culture of each microalgal species cryopreserved with different cryoprotectantagents and cooling methods for six months (I and III, kept in -196°C directly; II and IV, kept in -196°C after pre-cooling(-1°C min-1) to -80°C. No living cell was found on the final culture day. A different superscripted lower case letter in the samerow means a significant difference at the p < 0.05 level)
Growth of each microalgal species cryopreserved with different cryopreservative agents and cooling methods for six months (M, Me2SO; G, Glycerol; -196, kept in -196℃ directly; -80, kept in -196℃ after keeping in -20℃ and -80℃ for 10 min, respectively).
growth rate in a non-concentrated sample with the Me2SO (p < 0.05). Removing 10% Me2SO after thawing had no influence on the growth of the cells in a concentrated sample. P. tricornutum grew the best significantly in a non-concentration sample with the Me2SO removed (p < 0.05). The growth rate of S. costatum was not influenced by the removal of the Me2SO.
N. oculata grew better in a non-centrifugation sampleregardless of whether the Me2SO was removed or not.But S. maxima grew better with Me2SO than without itregardless of the concentration. N. oceanica showed thehighest growth rate significantly in a non-concentratedsample with the Me2SO removed (p < 0.05). Of theTetraselmis, T. striata grew better in a non-concentrationsample with the Me2SO in place. Only the growth rate ofT. suecica was not influenced by cell concentration andthe Me2SO. I. galbana grew better in a non-concentratedsample than a concentrated one regardless of whetherMe2SO was present or not. But P. pinguis, grew better ina concentrated sample with the Me2SO removed.
Freeze-drying is generally considered a better techniquethan serial transference, and the equipmentrequired is minimal. Freeze-drying, however, is unreliable,often affording survival rates less than 5% (Mcgrathet al. 1978). Cryopreservation is a method that has significantpotential for ensuring the long-term conservationand genetic stability of microalgal cultures (Poncet andVeron 2003). Many factors affect the effectiveness of cryopreservationin microalgal. But, one of the most importantfactors is the composition of the freezing medium.Although a good survival of deep-frozen microbes has
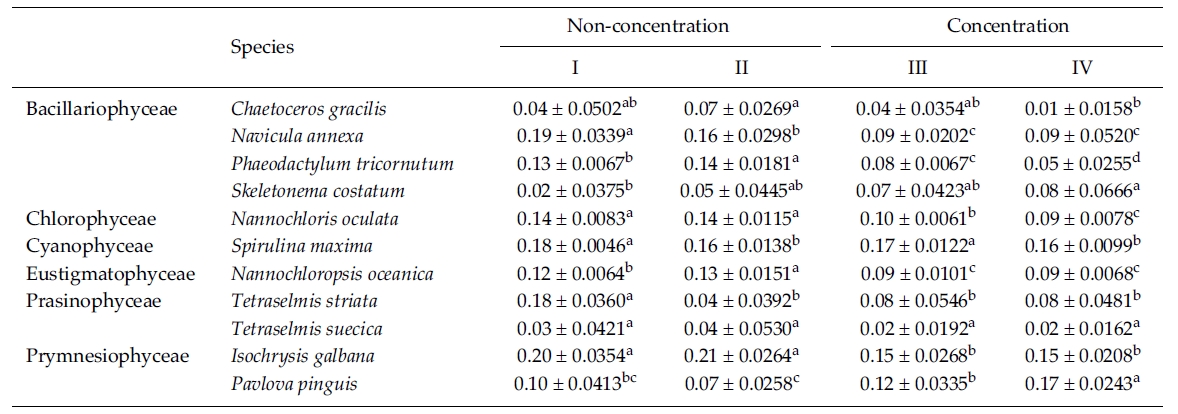
Mean daily specific growth rate for two months of each microalgal species cryopreserved for one year with different cryopreservationconcentrations and using the removal of Me2SO after thawing culture method (I and III, non-removal of Me2SOafter thawing; II and IV, removal of Me2SO after thawing. A different superscripted lower case letter in the same row means asignificant difference at the p < 0.05 level)
occasionally been observed without a protective additive,the presence of a suitable CPA usually increases thesurvival considerably (Hubalek 2003). The most commonlyused cryoprotectants in microalgae cryopreservationinclude: Me2SO, glycerol, methanol (MeOH), proline,polyvinylpyrrolidone and sorbitol (Canavate andLubian 1995a). However, CPAs are cytotoxic more orless. It is necessary to determine the tolerance of cells tothese compounds, especially when using the compoundsthat penetrate the cells such as glycerol and Me2SO.Fenwick and Day (1992) reported the effect of differentCPAs on cell motility in Tetraselmis suecica and glyceroland Me2SO were the least toxic. Prolonged exposure tothese cryoprotectants can actually harm the cells, soequilibration time and concentration of the cryoprotectantare important factors to control.
Cryoprotectant concentrations vary from 1 to 30%(v/v) in the alga culture media, but 5-10% solutions aremost common. However, survival of cryopreservedalgae largely depends on the degree of damage incurredduring the freezing and thawing process (Fenwick andDay 1992). Houdan et al. (2005) demonstrates that Me2SO(7.5%) could efficiently be used as a cryoprotectant forboth stages of the coccolithophore Emiliania huxleyi ifsubjected to a controlled rate of cooling. HaptophytesChrysochromulina simplex, Prymnesium parvum, and P.parvum f. patelliferum survived in a cryopreservationsolution of 15% Me2SO, as did three dinoglagellates:Amphidinium carterae, A. trulla, and Gymnodinium simples(Rhodes et al. 2006).
There have been ongoing efforts to improve freezingmethods. Morris (1976, 1978) examines the viability of250 strains of cryopreserved Chlorococcales; and Ben-Amotz and Gilboa (1980a, 1980b) tests 12 strains ofmarine green algae. In general, the cooling method consistsof slowly cooling the cultures at the rate of about -1°C min-1 between - 25°C to -70°C, and then plungingthem into liquid nitrogen for storage (Ben-Amotz andGilboa 1980a, 1980b; Day et al. 1997; Taylor and Fletcher1999; Tzovenis et al. 2004).
In the cryopreservation experiment on Navicula subinflataby Redekar and Wagh (2000), the optimum concentrationand exposure times of ethylene glycol, Me2SOand methanol are 4 M and 30 minute, respectively. Aftercryopreservation for 0 day, 7 days and 30 days, the maximummicroalgae growth rate was recorded for ethyleneglycol but there was no growth in methanol.
Tetraselmis was successfully cryopreserved with 10 or15% Me2SO (Rhodes et al. 2006). According to Fenwickand Day (Fenwick and Day 1992), Tetraselmis is bettercryopreserved with glycerol than with Me2SO. For T. suecica(Fenwick and Day 1992; Montaini et al. 1995) glycerolhad the most protective effect with post-thaw viabilitylevels of over 70%. T. chuii was cooled at a rate of 0.5°Cmin-1 from 0°C to -50°C before being plunged into liquidnitrogen. T. chuii was successfully cryopreserved using5% Me2SO for 3-5 weeks (Canavate and Lubian 1997a).In this study, T. suecica was also successfully cryopreservedwith rapid cooling using 10% glycerol.
Nannochloris oculata displayed different levels of postthaw viability depending on the CPAs (Poncet and Veron 2003). Microalgae cryopreserved with methanol did not grow for the first six days. In contrast, that of theunfrozen methanol-treated control cultures increasedover this period. After six days in culture, the growth ofthe cryopreserved microalgae subsequently recovered,but cell density at the end of the experiment was lowerthan that of the control group. Gwo et al. (2005) finds thatthe viability of N. oculata is best when using a coolingrate of 1°C min-1 to -40°C and liquid nitrogen with theaddition of either Me2SO or propylene glycol at a 10 or20% concentration. N. oculata was successfully cryopreservedusing Me2SO 2.5 M and 2.2 M glycerol. The viabilityrate of N. gaditana, which was cryoperseved with5% methanol for seven days, was 49% (Canavate andLubian 1995b). The cryopreservation of this microalgalspecies at the lowest cooling rate (0.5°C min-1) in a mediumof 20 ppt salinity showed higher viability regardlessof whether Me2SO was added or not (Canavate andLubian 1997a).
After being frozen using a rapid uncontrolled coolingmethod - a mechanical freezer set at -60°C, - bloom-formingcyanobacterial strains such as Microcystis,Oscillatoria, Anabaena, and Aphanizomenon grew sufficientlyfor the test of post-thaw viability. Complete viabilityof all the tested strains was found after 2.5 years ofcryopreservation without cryoprotectant (Park 2006).
Many previous studies have considered Me2SO as oneof the best CPAs for microalgae (Taylor and Fletcher1999). Me2SO has been used successfully to preservemicroalgae at concentrations ranging from 3% v/v forthe freshwater cyanobacterium Microcystis aeruginosa to15% for several marine species belonging to differentPhyla (Canavate and Lubian 1995a). However, in thisstudy, O. angustissima was successfully cryopreservedusing 10% Me2SO. The effect of slow and rapid coolingon the viability of O. angustissima was different inMe2SO.
On the other hand, new technique on encapsulationand dehydration of cryopreserved cell has recentlydeveloped to reduce the mortality of the microalgae dueto freezing. However, the results of this technique up tonow are also variable according to microalgal species(Zhang et al. 2009). Using this technique, the viability ofNannochloropsis sp., Nostoc commune and Phormidium foveolarumwere successfully recovered after storage in liquidnitrogen. But that of Spirulina subsalsa, Volvox aureus,Pandorina morum and Rhodomonas ovalis did not grow(Hirata et al. 1996; Day et al. 2000).
Many results on microalgae cryopreservation techniqueshave been reported. Nevertheless, only a fractionof the large number of microalgae kept in culture collectionshave been successfully regenerated after storage inliquid nitrogen (15% of the National institute forEnvironmental Studies in Japan, 35% of CultureCollection of Algae and Protozoa in the United Kingdom(Day et al. 1998), and 40% of the Provasoli-GuillardNational Center for Culture of Marine Phytoplankton inthe U.S.A, ccmp.bigelow.org, 2009).
In this study, microalgal species with thick cell wallswithout flagellates or seta were successfully cryopreserved.When compared with glycerol, Me2SO provedthe most efficacious for cryopreservation. The cell concentrationand amount of time in cryopreservation hadno affect on the viability and growth of the cell. In addition,growth of microalgae after thawing was not influencedby the removal of the 10% Me2SO. The age of a cellculture was also considered as an important factor indetermining its viability. The resistance of cells to thestress of freezing and thawing increases at the stationaryphase of growth because cells in this physiological stateaccumulate lipid content (Harding et al. 2004). The viabilityand growth of cryopreserved microalgae depends onthe species. There is unlikely to be a uniform protocolthat could be applied to various circumstances. The useof cryopreservation at the KMMCC is still at a preliminaryphase of investigation, although a significant numberof microalgal species has been successfully cryopreserved.It is difficult to infer the best cryopreservationmethod for microalgal species. Cryopreservation affectsdifferent species differently. Therefore, the optimumcondition for successful cryopreservation should beexamined for each microalgal species.