Stains from medicinal plants may be used to define and examine bulk tissue and cell populations and to classify different blood cells or organelles within individual cells [1]. The medicinal plant henna produces a red orange dye molecule, which is known as hennotannic acid (lawsone), which has an affinity for bonding with proteins and has been used to dye skin, fingernails, hair, leather, silk and wool [2].
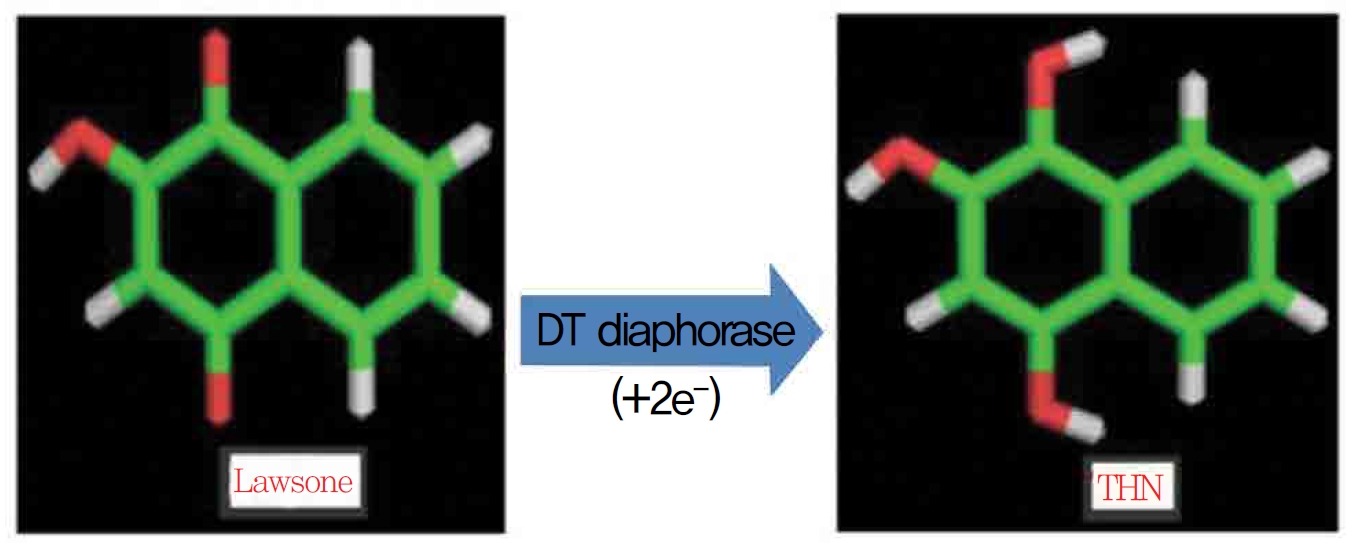
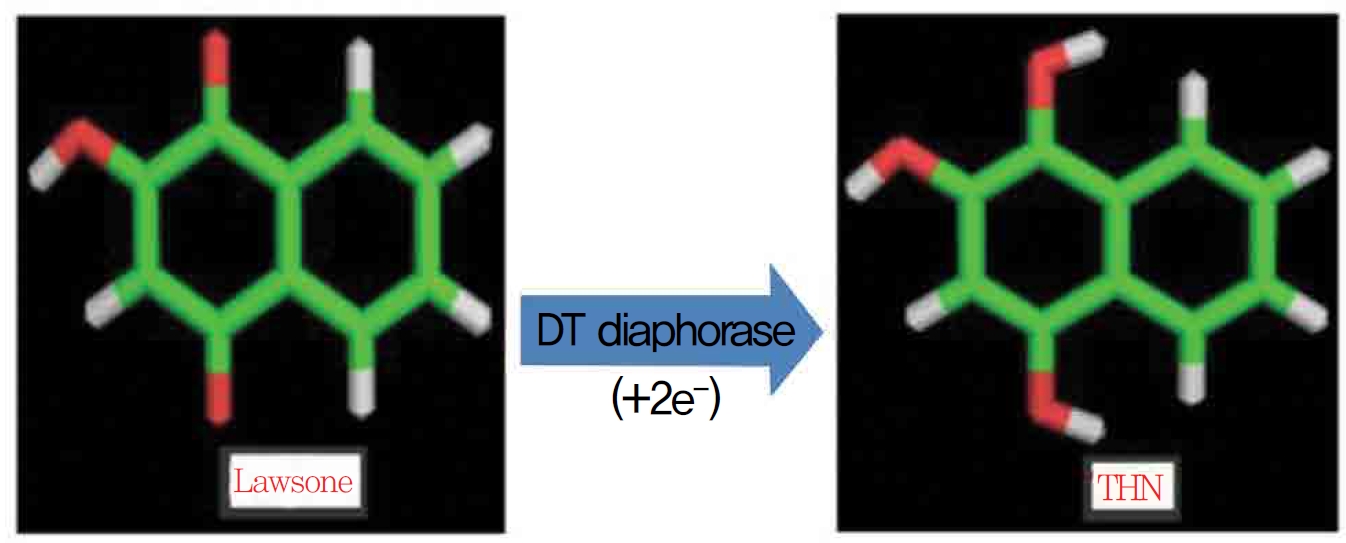
1,4-naphthoquinone (lawsone) is able to induce oxidative damages as a consequence of its ability to undergo redox cycling and to generate reactive oxygen species. Lawsone (2-hydroxy 1,4-napththoquinone) is an active naphthoquinone derivative and a non redox cycling compound. The apparent lack of oxidative activity of lawsone has been explained by the stabilizing influence of its acidic 2-hydroxy group, which is thought to impede its ability to redox cycle (Fig. 1) Methylation of the 2-hydroxy group yields a quinine with a much greater ability to redox cycle. Some suggest the involvement of a superoxide lawsone, and oxidative stress probably arises when the naphthoquinone part in lawsone is reduced to a semi quinone by enzymatic systems. DT-diaphorase (DTD) is known to catalast a two electron reduction of lawsone to its hydroquinone, 1,2,4 trihydroxynaphthalene (THN).
Previous studies on lawsone have had conflicting results in that lawsone cannot generate reactive oxygen species at a rate sufficient to exceed the capacity of the cells to remove them by nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) quinine reductase, which eliminates lawsone toxicity through the kidneys. The electron acceptor specificity of DTD is very similar to that of NADPH quinine reductase, and induction of this enzyme response minimizes the toxicity of lawsone. The scientific committee on cosmetic products and non food products intended for consumers of the European Union concluded that lawsone had mutogenecic/genotoxicity and considered banning lawsone for the use as a non oxidizing hair dye, as well as for other uses [3]. Repeated doses of lawsone can provoke an anemic response with a genetic deficiency in erythrocytic glucose 6-phosphate dehydrogenase (G6PDH) activity and organs damage [4]. Some studies have concluded that lawsone is mutagenic, but not others [5].
Prediction of the toxicities of the ligands on proteins plays a major role in drug discovery through in-silico modeling using different approaches. Due to the toxic and the non toxic properties of lawsone, in the present study, a preliminary molecular docking analysis of lawsone and its metabolite THN was done by using AutoDock docking tool 4.0. Proteins such as hydrogen peroxide (H2O2), nitric oxide synthase (NOS), catalase (CAT), glutathione (GSH), glutathione reductase (GR), G6PDH and NADPH were subjected to energy optimization by adding solvation effects for predicting the binding affinities of lawsone and THN ligand molecules for their respective receptor binding sites.
Protein data bank (PDB) chemical structures of the ligands lawsone and THN and of the proteins receptors H2O2, NOS, CAT, GSH, GR, G6PDH and NADPH were used in this study. AutoDock 4.0 and python molecular viewer were used for the docking analysis. The 3D structures of the ligands were drawn using hyperchem drawing tools and were then saved as a pdb file. Energy minimization of all pdb files was carried out with the help of hyperchem by MM+ and by using a semi-empirical (PM3) method. The proteins models were derived with the help of a pbd online search. The ligands and the proteins were taken for the docking procedure, and the docking studies were done by using the program AutoDock 4. The docking process begins with a ligand molecule so that suitable dockings at the protein binding sites can be identified through annealing and genetic simulation algorithms.
The program auto dock tools (ADT) released as an extension suite to the python molecular viewer was used to prepare the proteins and the ligands. For the macromolecules downloaded from the PDB server [6], polar hydrogen was added; then kollman united atom charges and atomic solvation parameters were assigned. The grid maps of the docking studies were computed using the Auto Grid 4.0 included in the AutoDock 4.0 distributions. Grids centered on active sites were obtained by using spacing grid size of 54 × 55 × 56, with a calculated grid spacing of 0.503. The GA-local search (GA-LS, A Comparison of Global and Local Search Methods in Drug Docking) method was adopted to perform the molecular docking. The parameters for the GA-LS were defined as follows: a maximum number of 2,500,000 energy evaluations, a maximum number of generations of 27,000, and mutation and crossover rates of 0.02 and 0.8, respectively. The number of docking runs was set to 10. After docking, structures generated were assigned to clusters based on a tolerance of 1 Å for the root means square deviation (RMSD) from the lowest energy structure. The binding energy, ligand efficiency, intermolecular force, and van der Waal interactions between docked potent agents and macromolecules were analyzed using ADT (version 1.5.4).
In the present study, the three dimensional structures of H2O2, NOS, CAT, GSH, GR, G6PDH and NADPH were retrieved from the PDB site [6]. Ligands were docked with the candidate proteins by forming grids and were saved in docking log file (dlg) format in AutoDock 1.5.4. The docking calculation in Auto Dock was performed by using the receptor protein and ligands, and the results were viewed by reading the docking log file (dlg) in the analyze menu. The dlg file was opened, and different conformations were loaded in increasing order of energy; then, the play option was selected to visualize the conformation. The docking properties were checked, and the energy optimized ligand was subjected to a docking analysis to check for its efficiency as an agonist of proteins. Similarly, the docking calculator was set for proteins with lawsone and THN, and the best binding modes were analyzed (Table 1).
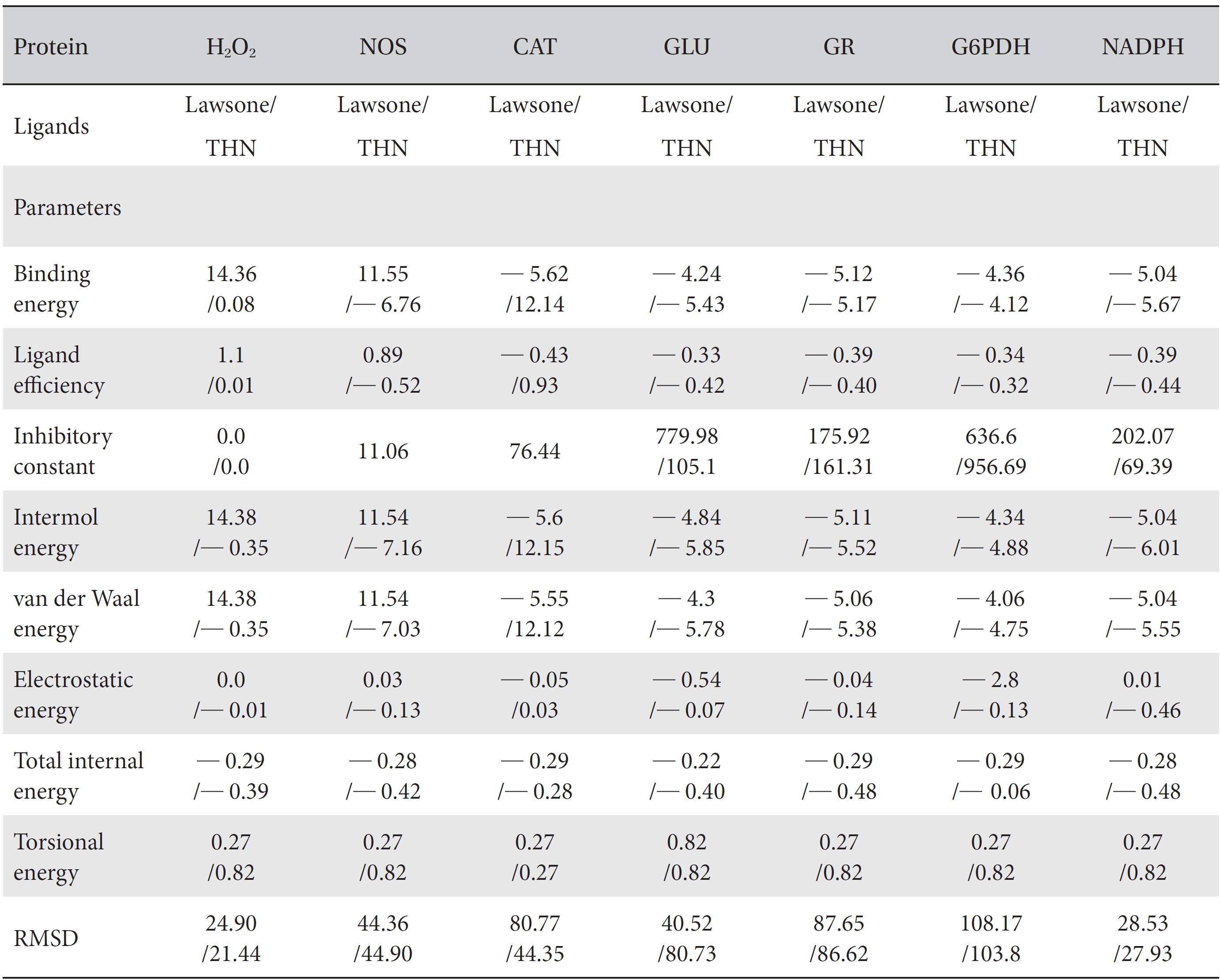
[Table. 1] Comparison of the protein binding energies and other parameters of lawsone and THN
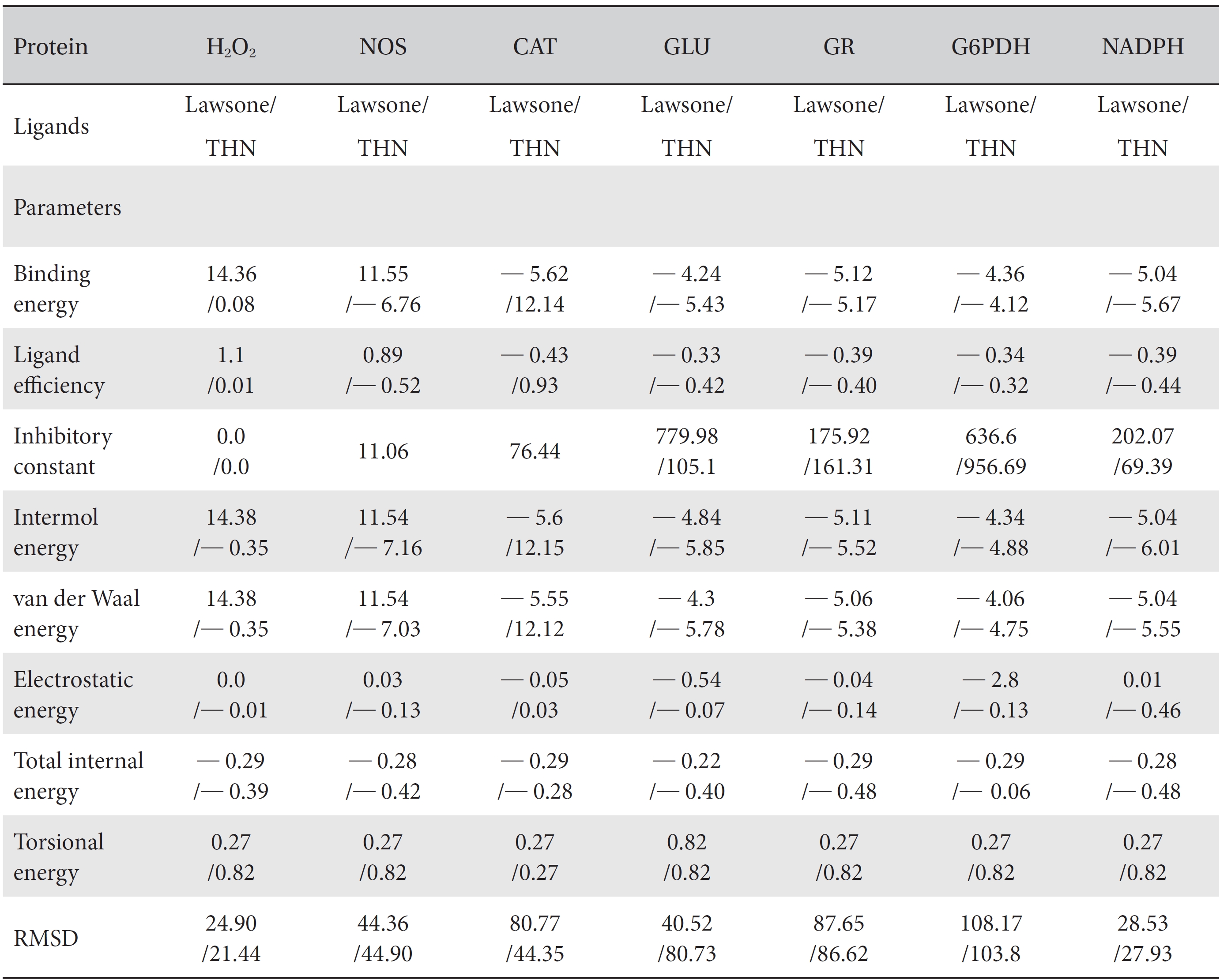
Comparison of the protein binding energies and other parameters of lawsone and THN
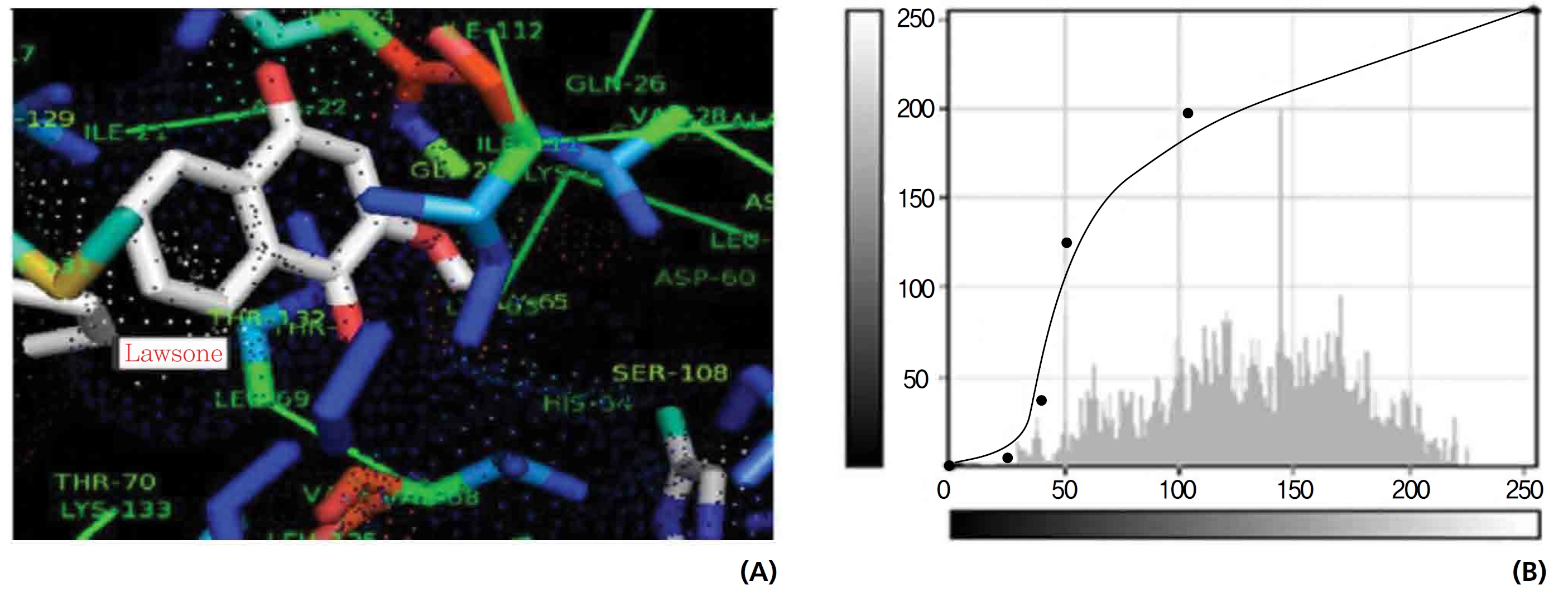
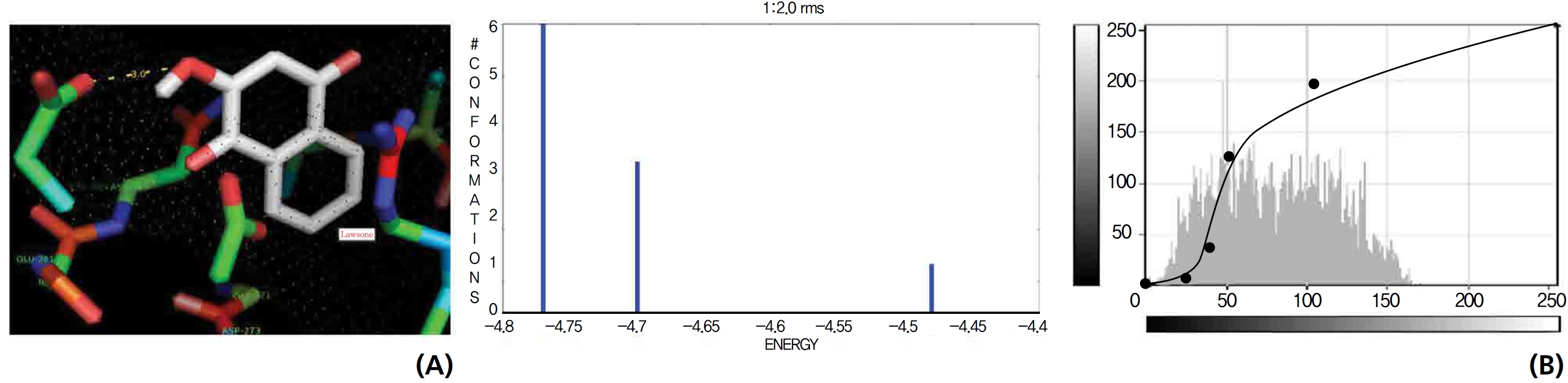
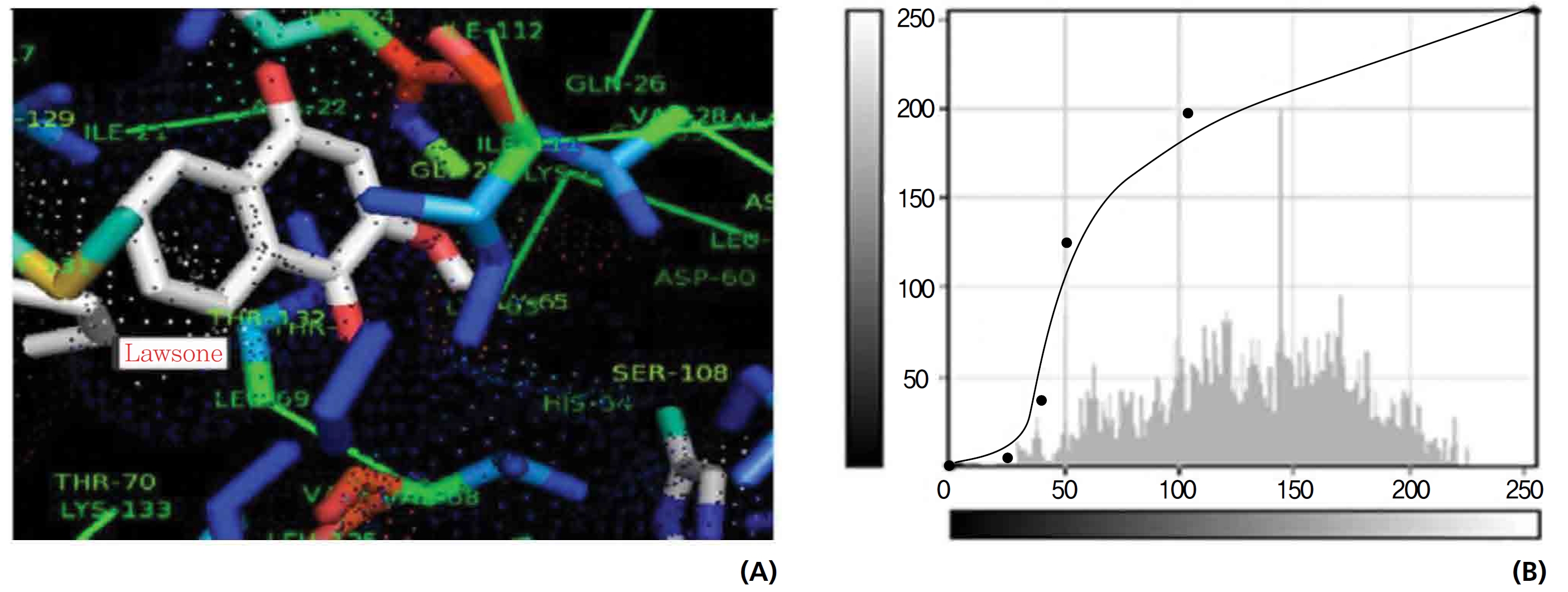
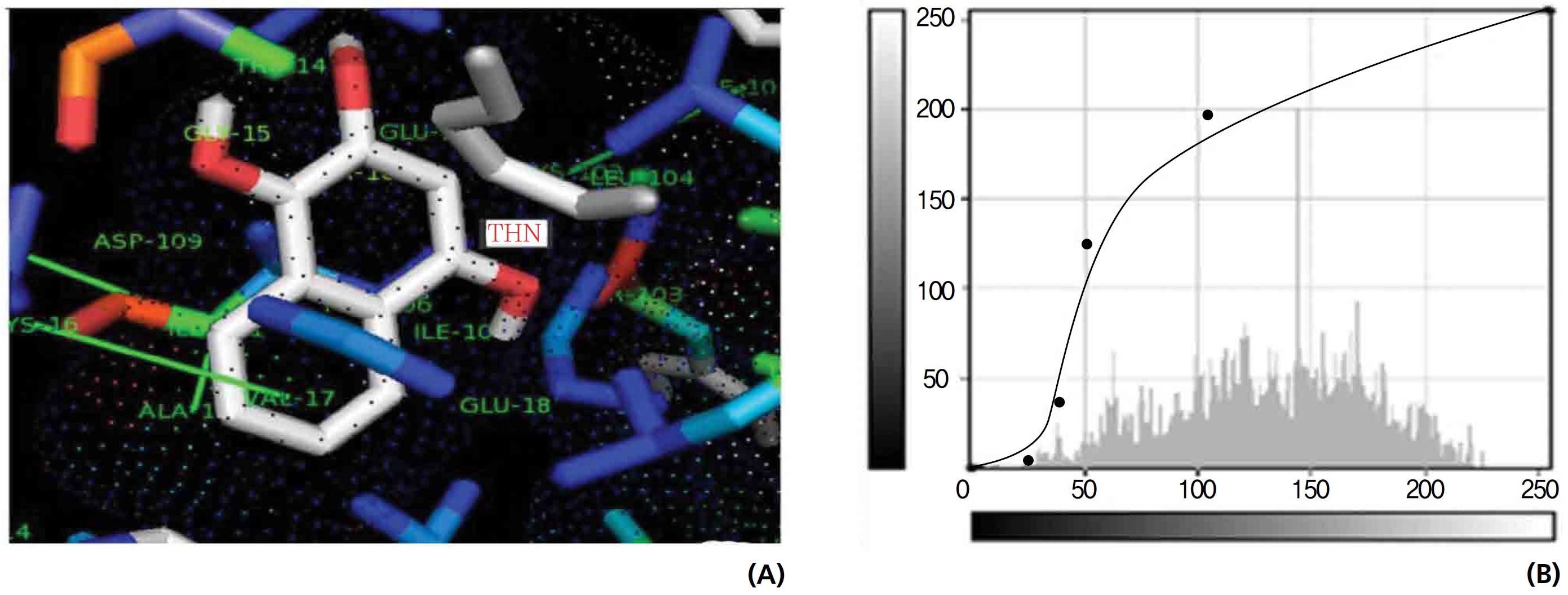
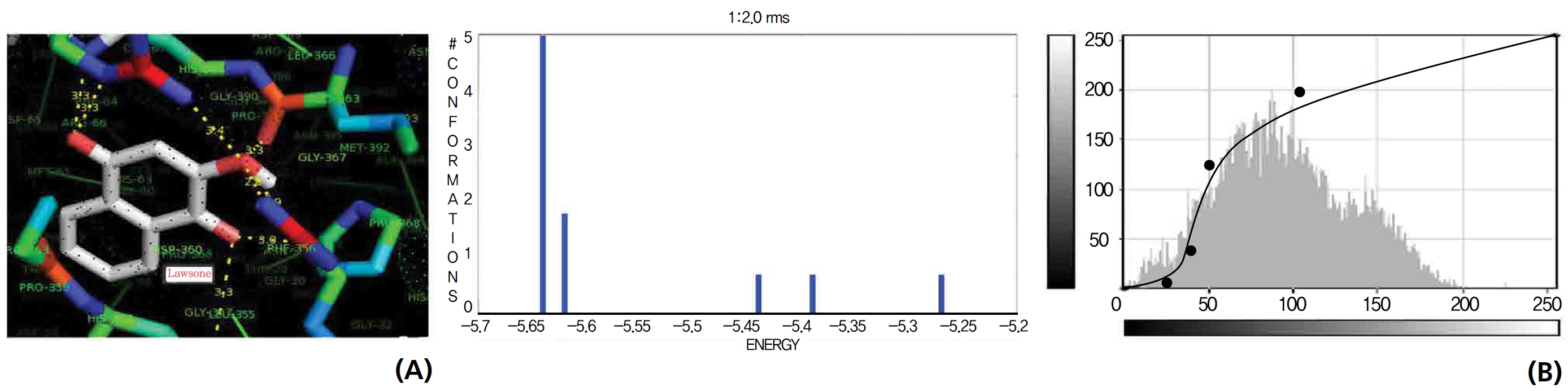
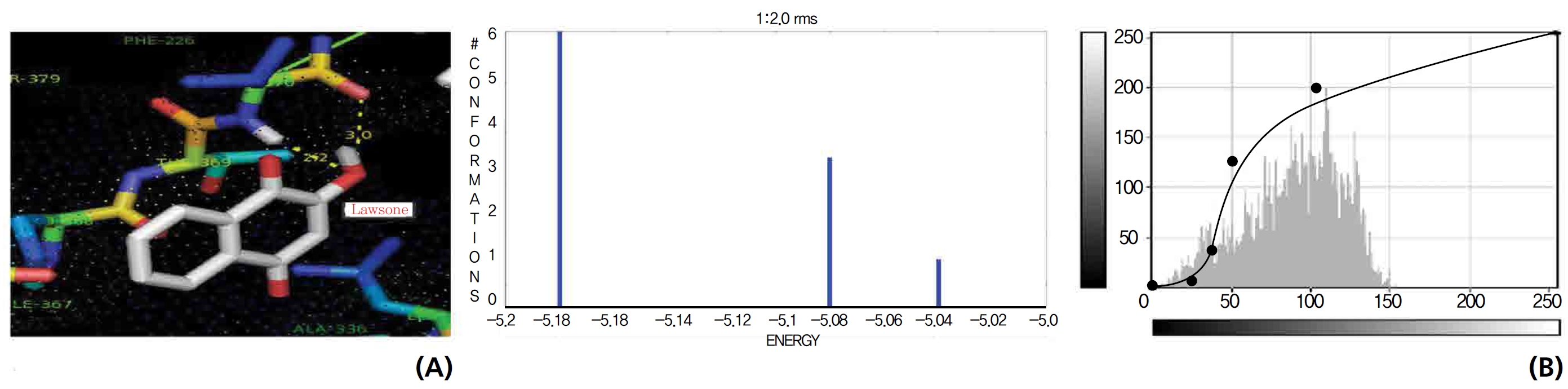
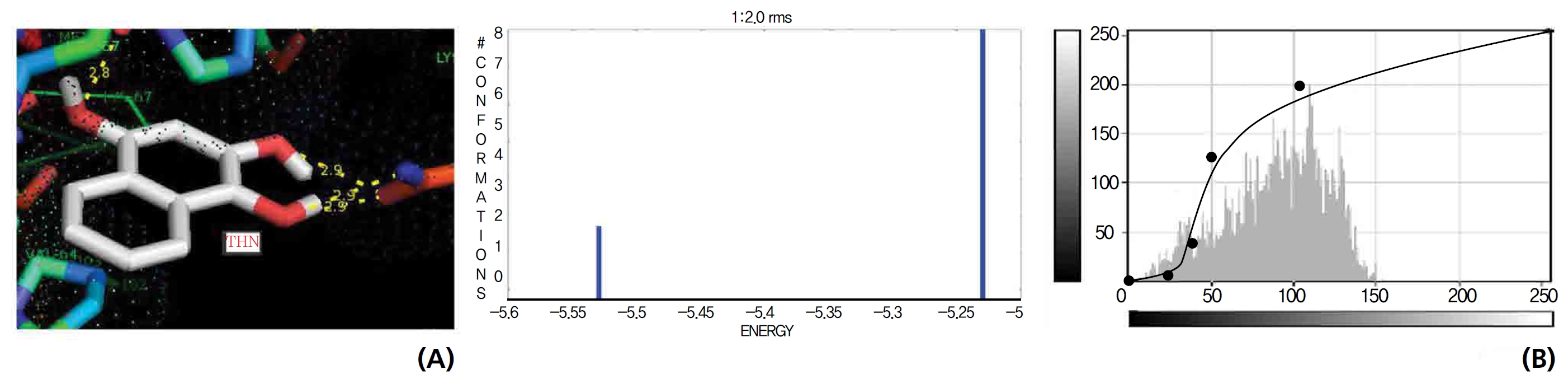
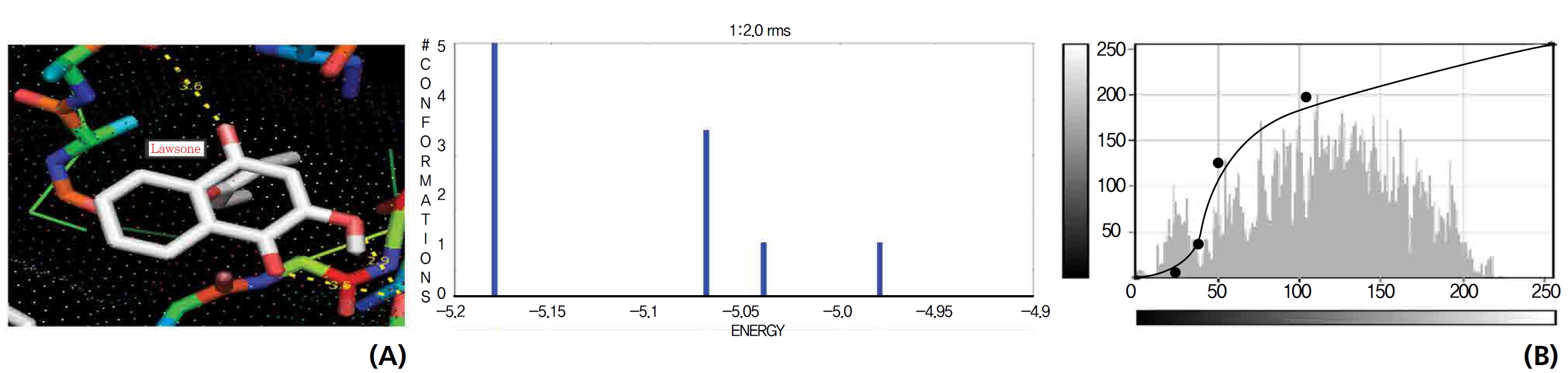
On the basis of molecular docking, the H2O2 binding interactions of lawsone and THN were evaluated. This study showed that lawsone and THN did not have interactions with H2O2 to any great extent, which was confirmed by the positive values for the binding energies, which were 14.36 for lawsone and 0.08 for THN. The results are presented in the Figs. 2,3; none of the catalytic residues took part in these interactions.
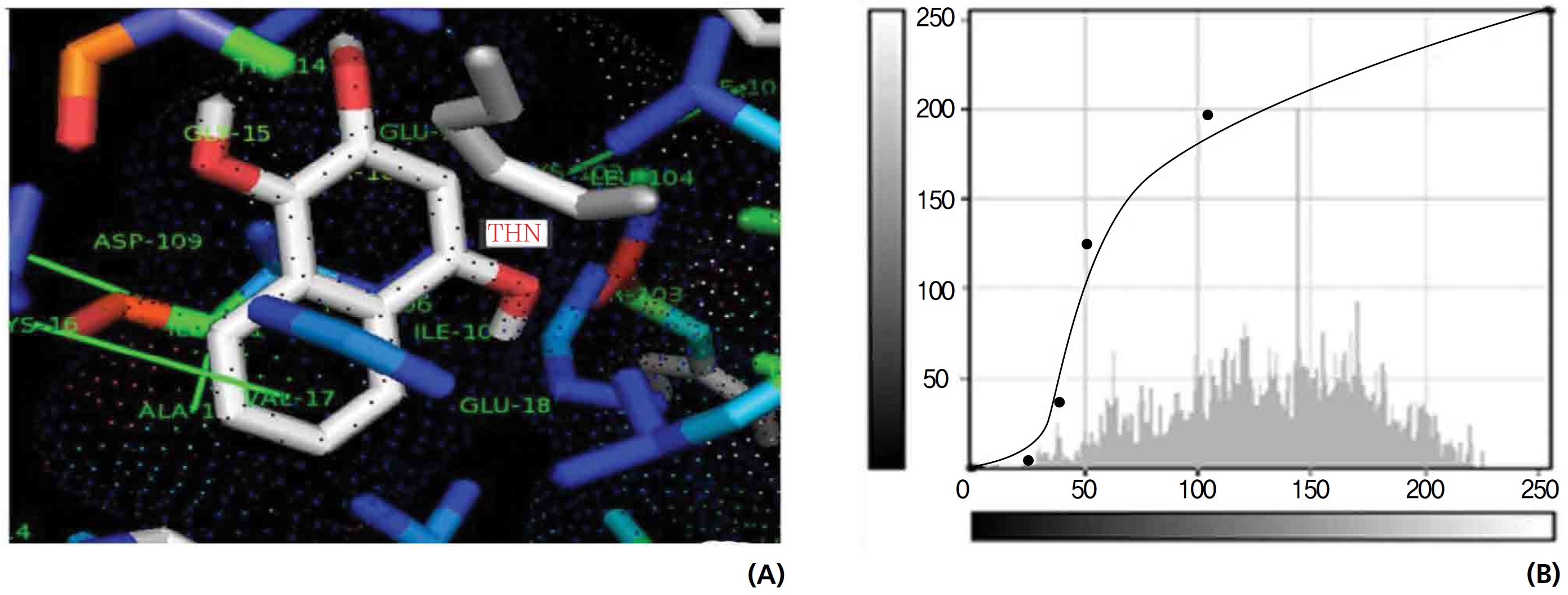
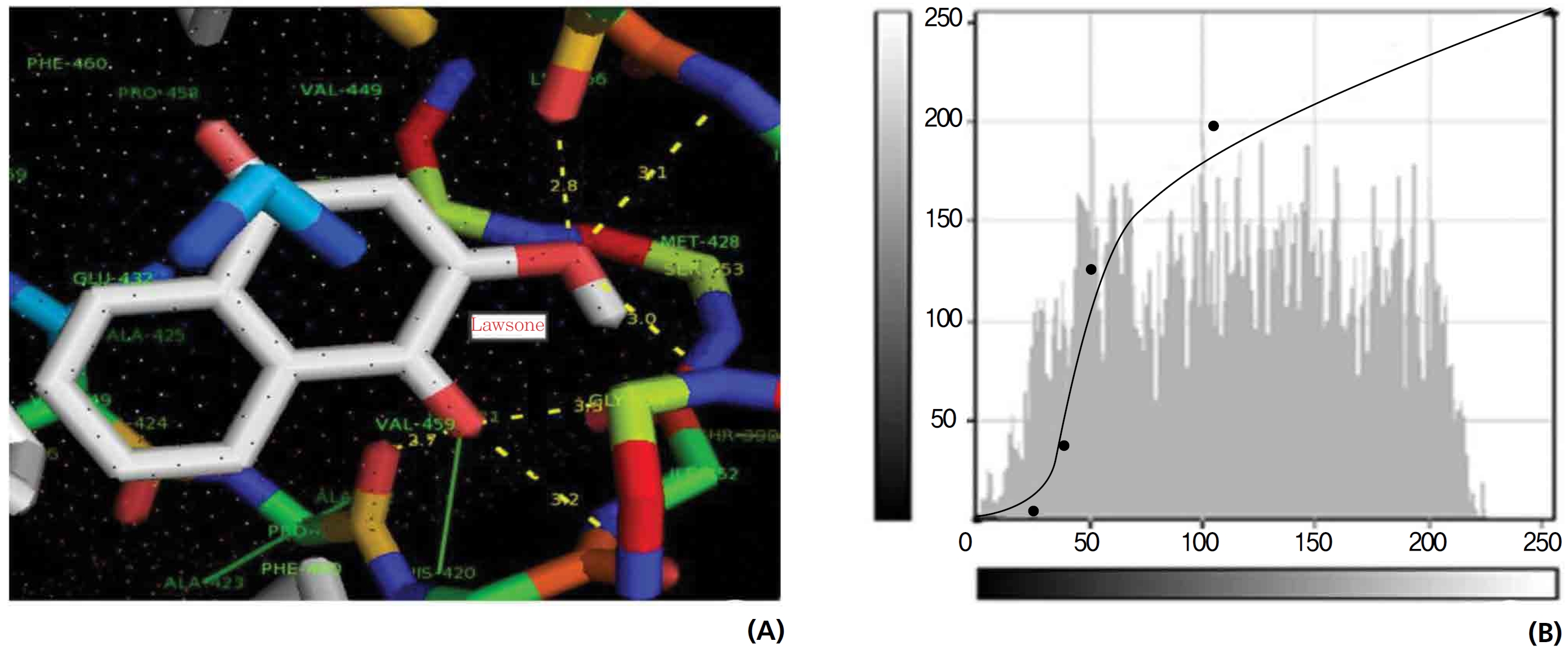
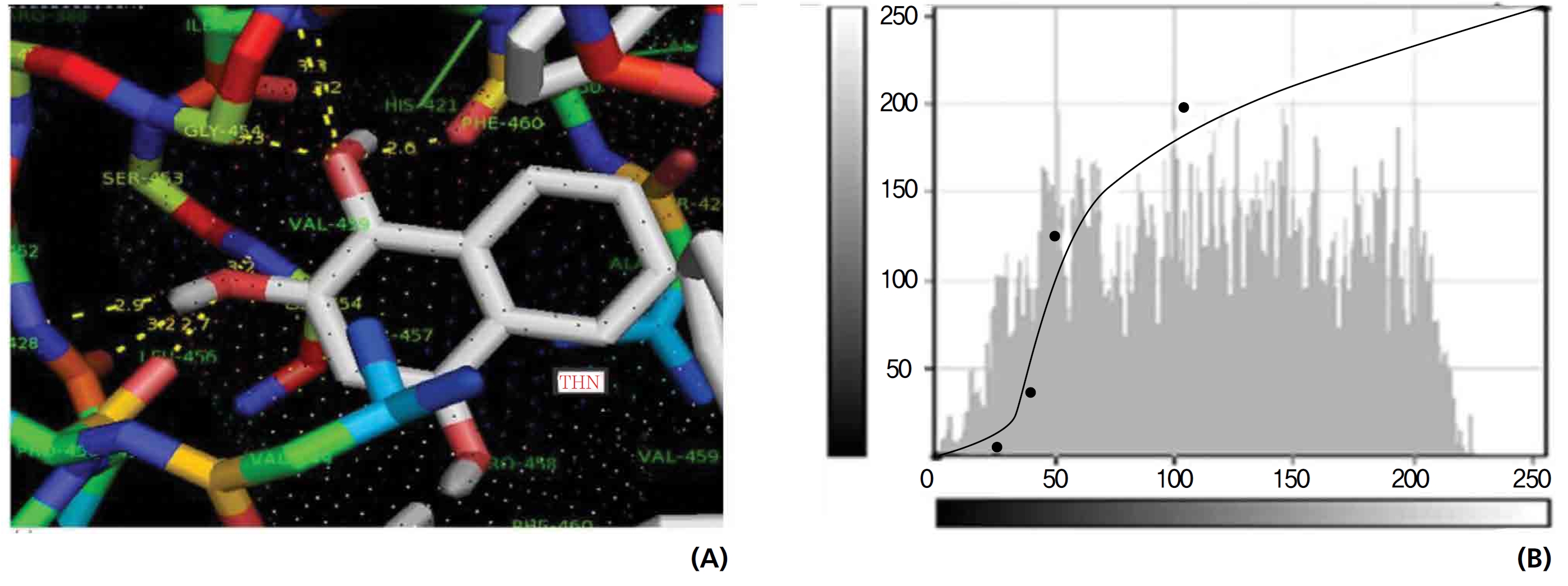
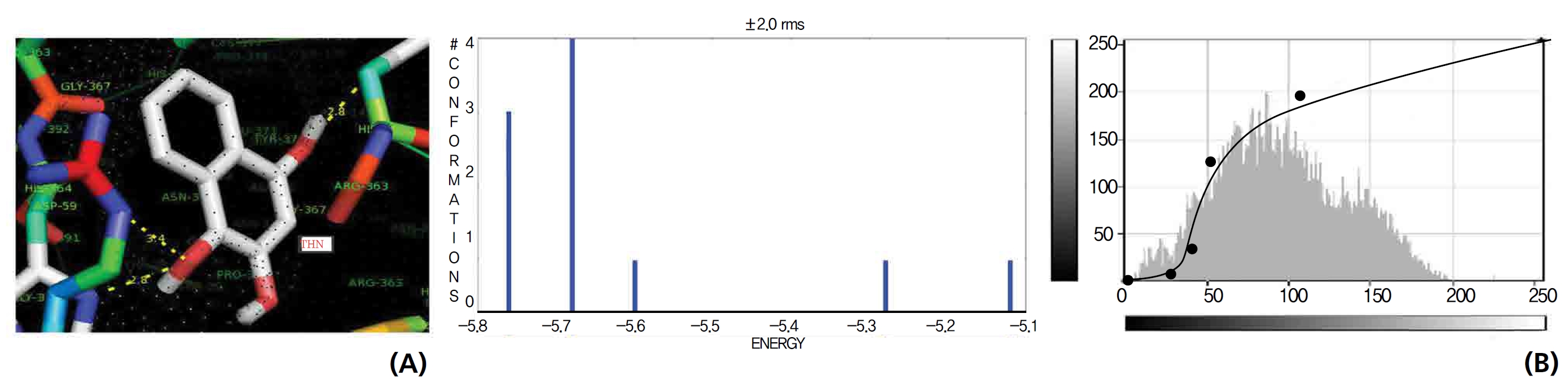
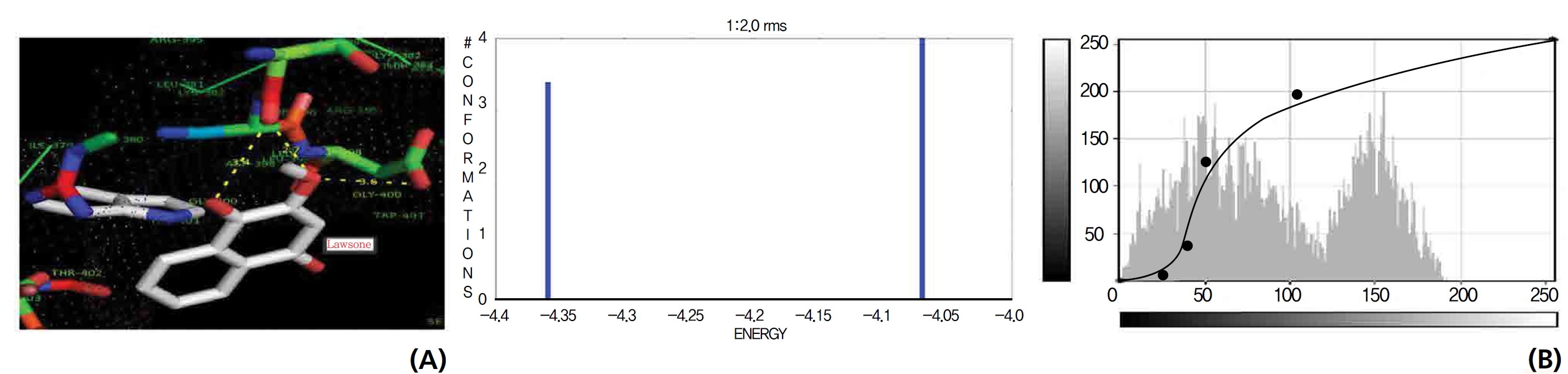
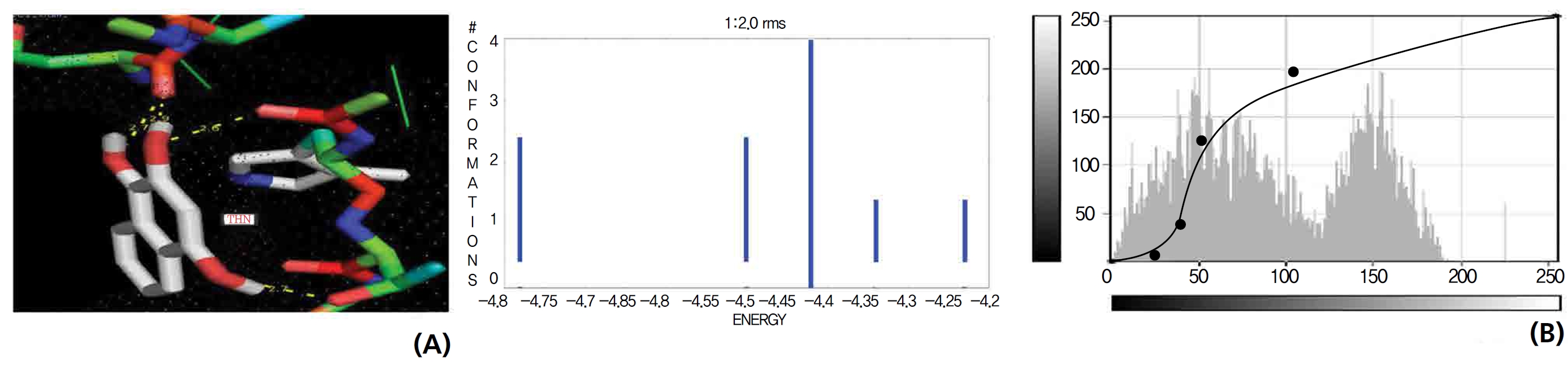
The NOS protein was chosen, and the docking binding energy of lawsone energy for NOS was found to be 11.55 kcal/mol-1; that for THN was found to be ─ 6.76 kcal/mol-1. The latter result was confirmed through the major docking parameters: an intermolecular energy of ─ 7.16, a van der Wall force of ─ 7.03, an electrostatic energy of ─ 0.13, and a total internal energy 0.42, with all values being in units of kcal/mol-1. The positional affinities of one hydroxy (OH) of THN was identified for a proline oxygen atom, a leucine nitrogen atom, and a glycine nitrogen atom 3.3 kcal/mol-1, 3.2 kcal/mol-1 and 3.3 kcal/mol-1, respectively. The 2-hydroxy of THN shows amino acid sites that have an affinities for glycine nitrogen, proline oxygen and leucine oxygen: 2.7, 2.9, 3.2 and 3.3 kcal/mol-1, respectively (Figs. 4,5).
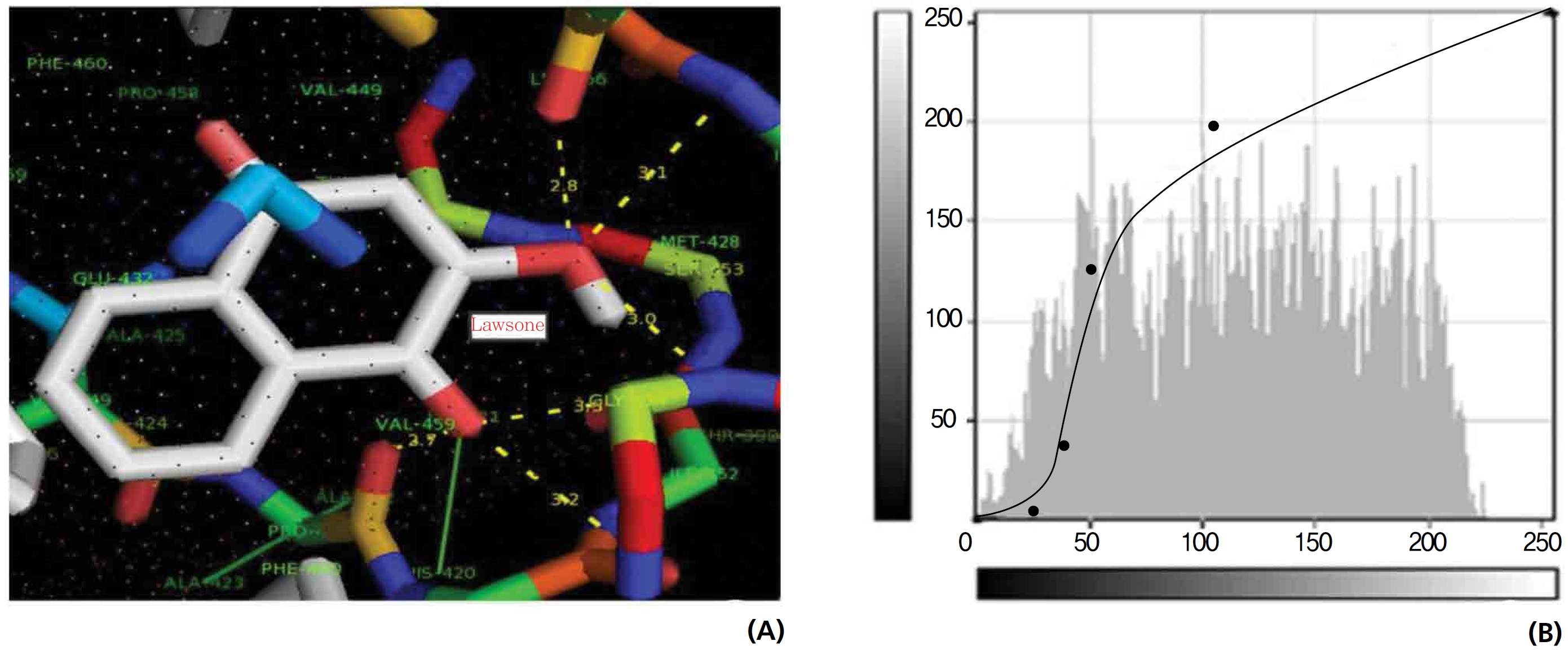
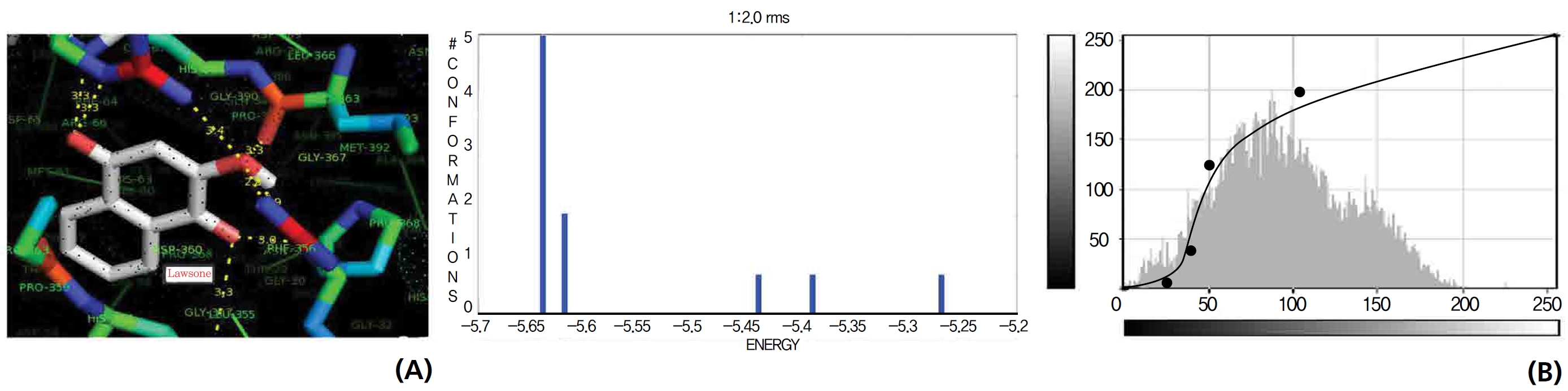
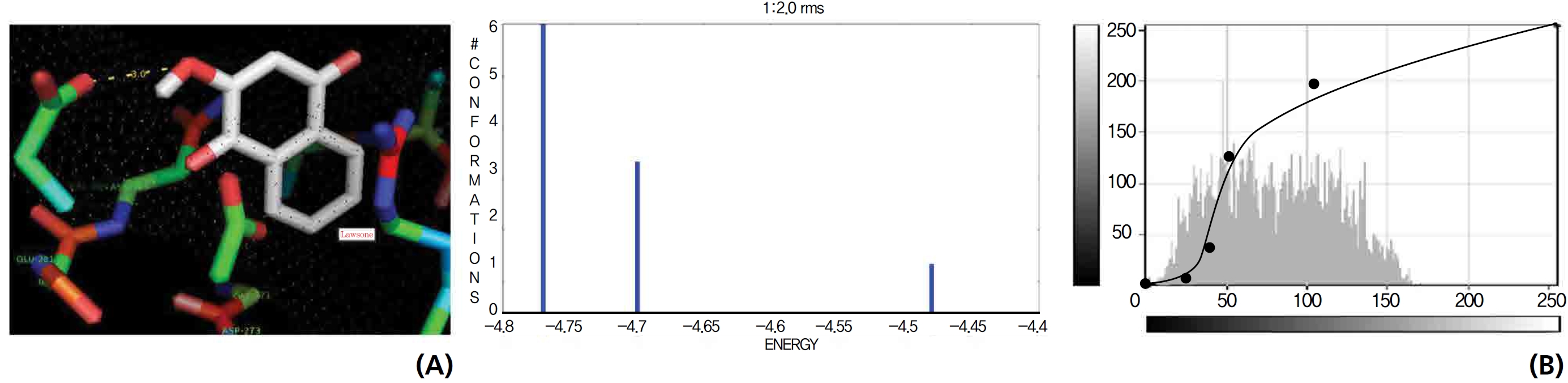
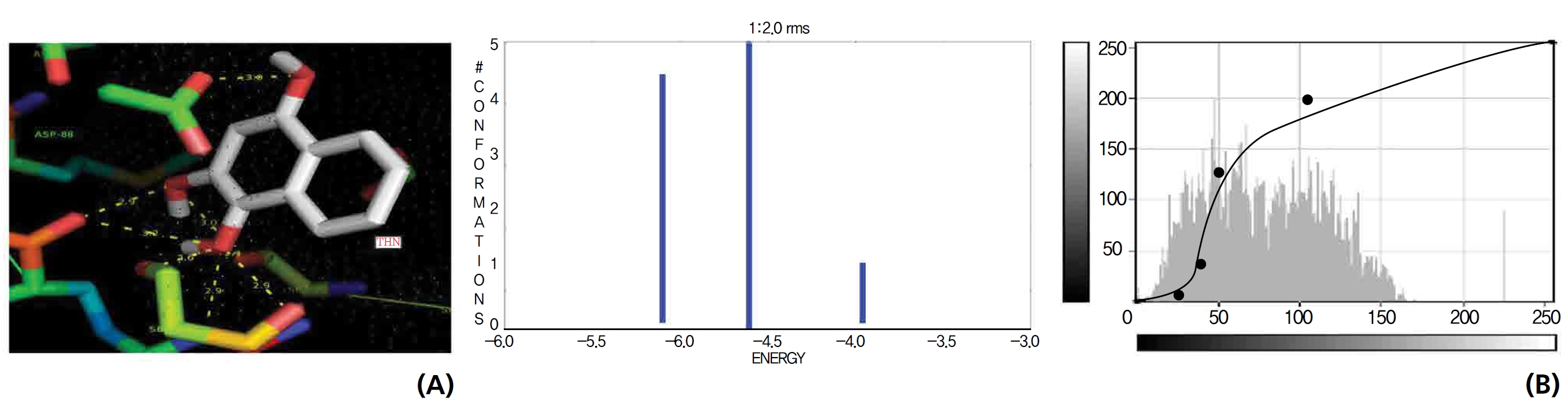
A protein catalase was selected for lawsone and THN molecular binding. The study result showing a better affinity of lawsone for the CAT interaction was confirmed through binding energy, ligand efficiency, intermolecular energy, van der Waal energy, electrostatic energy and total intermolecular energy measurements. Fig. 6 presents the hydrogen intermolecular forces of lawsone with alanine and arginine through oxygen and nitrogen atoms at 2.8, 2.9, 3.3 and 3.4 kcal/mol-1, respectively. The hydrogen atom free force for one oxy (O) position of lawsone with amino acid residues such as alanine (through oxygen) and thrionine (through nitrogen) were evaluated as 3.0 and 3.3 kcal/mol-1, respectively, and that of 4-O of lawsone with arginine and alanine amino acids through nitrogen was 3.3 kcal/ mol-1 (Figs. 6,7).
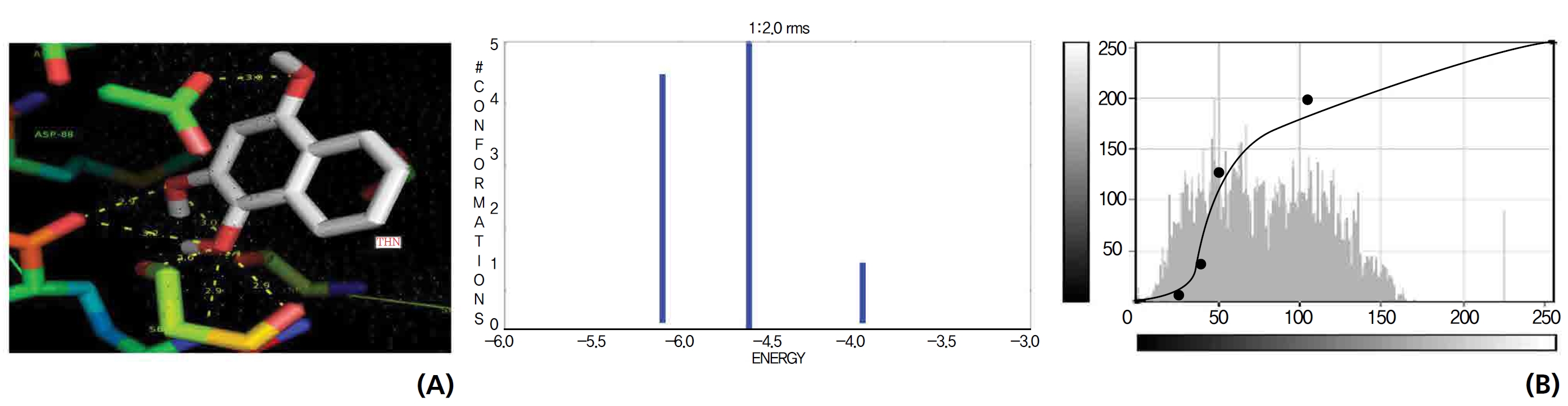
A docking study comparison was made between lawsone and THN for the GSH receptor interaction. Significant variations were observed in the affinity of THN for GSH with respect to binding energy (─ 5.43 kcal/mol-1), ligand efficiency (─ 0.42 kcal/mol-1), intermolecular energy (─ 5.85 kcal/mol-1), van der Waal force (5.78 kcal/mol-1), total intermolecular energy (─ 0.40 kcal/mol-1) and RMSD (80.74 kcal/mol-1), THN shows a dominant molecular interaction with the GSH receptor. Through the positional affinities of OH, 2-hydroxy (= OH) and 4-hydroxy (= OH) of THN for amino acids, their molecular affinities for aspartine, serine and arginine were predicted. The results are as follows: 1 OH with aspartine oxygen at 3.0 kcal/mol-1, 2-hydroxy with serine oxygen at 2.4, 2.6 and 3.0 kcal/mol-1, and 4-hydroxy with arginine oxygen and nitrogen at 2.9 and 3.2 kcal/mol-1, respectively. The results in Figs. 8,9 show that the additional amino acids interactions were found and analyzed for the 1-OH and 4-hydroxy (= OH) positions of THN with nitrogen atoms of aspartine (2.8 kcal/mol-1), thrionine (3.4 kcal/mol-1) and histidine (2.8 kcal/mol-1).
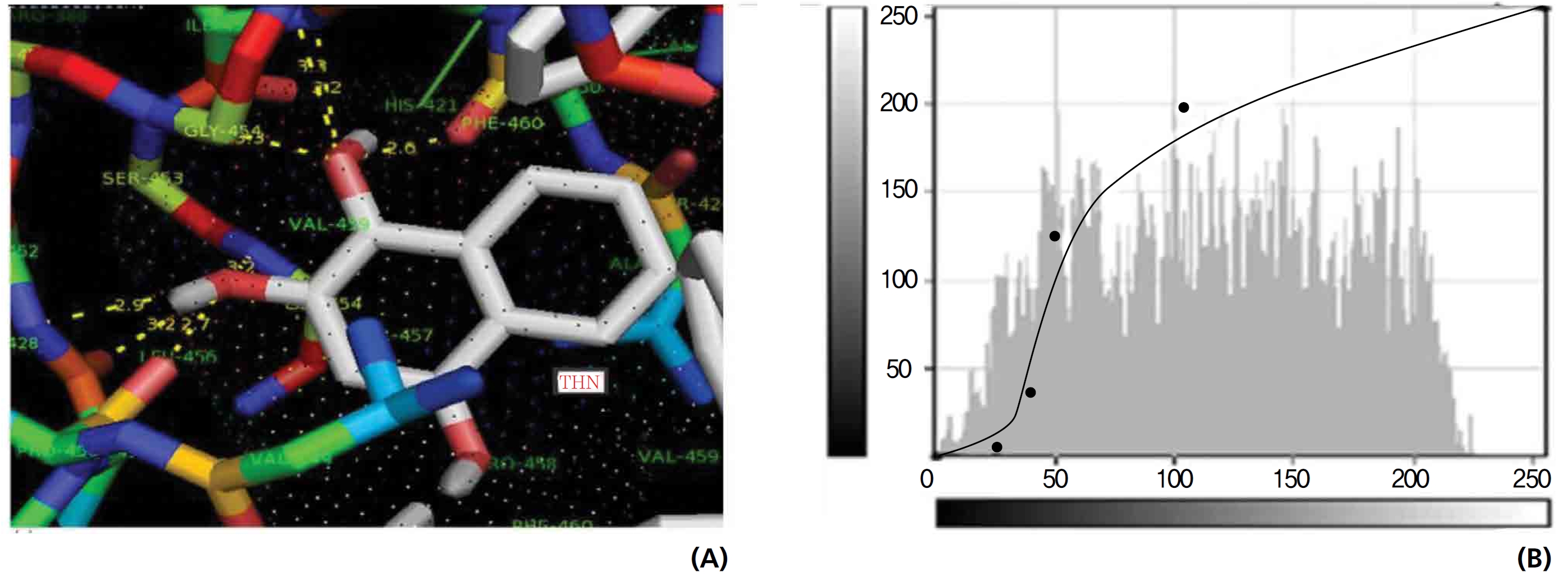
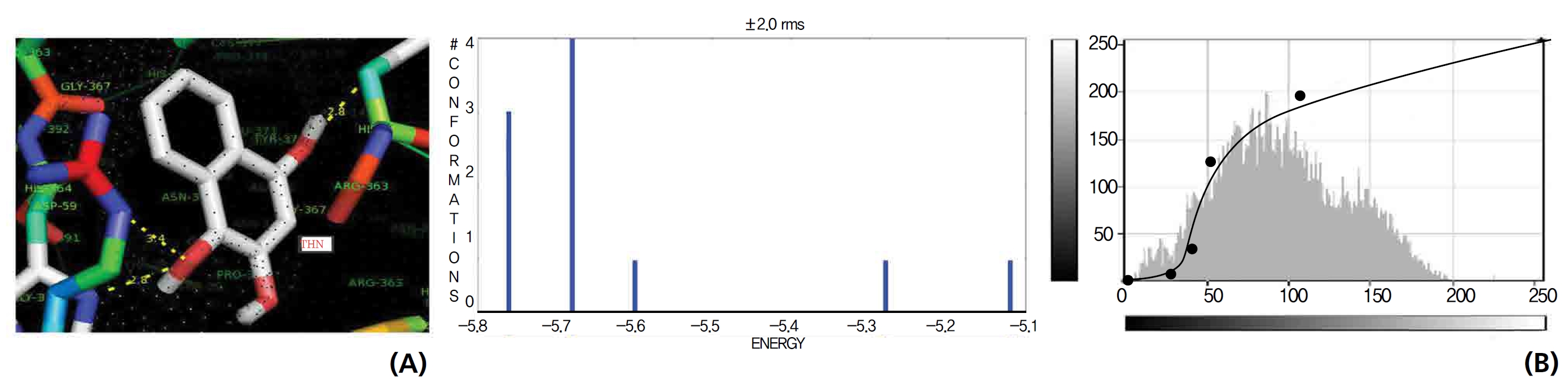
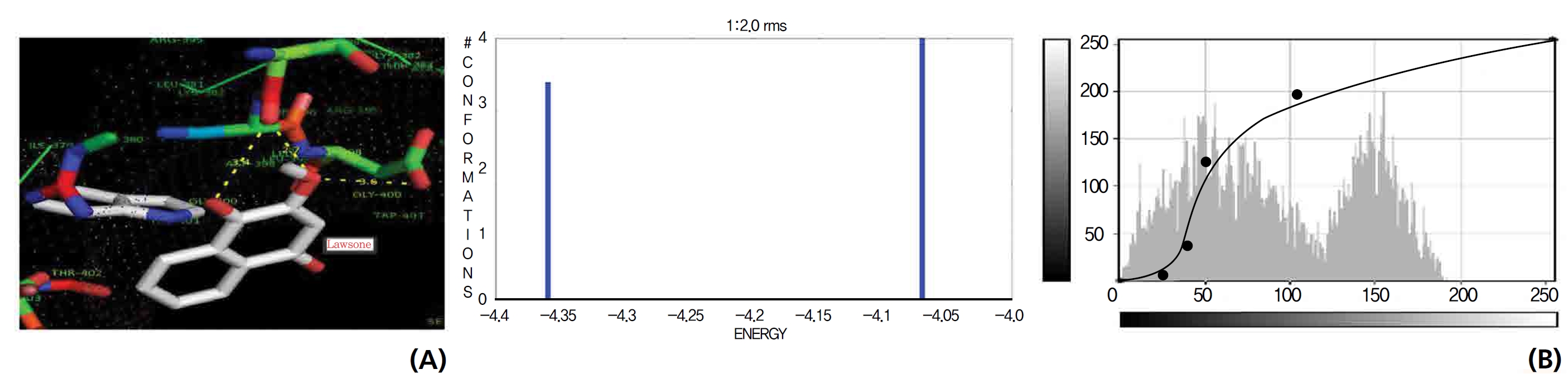
The GR interaction was investigated for another protein affinity evaluation for lawsone and THN. The four docking parameters, binding energy, ligand efficiency, intermolecular energy and van der Waal force showed almost one and the same interactions of lawsone and THN with protein GSH reductase: ─ 5.12, ─ 0.39, ─ 5.11, and ─ 5.06 kcal/ mol-1 for lawsone and ─ 5.17, ─ 0.40, ─ 5.52, and ─ 5.38 kcal/mol-1 for THN, respectively. The electrostatic energy and the total intermolecular energy showed the leading molecular interactions for THN rather than for lawsone: ─ 0.14 and ─ 0.48 kcal/mol-1, respectively. The 2-hydroxy position of lawsone interacted with the one mole oxygen of valine and one mole hydrogen of threonine: 2.2 and 3.0 kcal/mol-1, respectively. In the case of THN amino acid interactions, one-hydroxy had high affinities for the nitrogen and the oxygen of glycine (2.8 kcal/mol-1), two-hydroxy had a high affinity for the oxygen of glycine (2.9 kcal/mol-1), and four-hydroxyl had a high affinity for proline oxygen (2.8 kcal/mol-1) (Figs. 10,11).
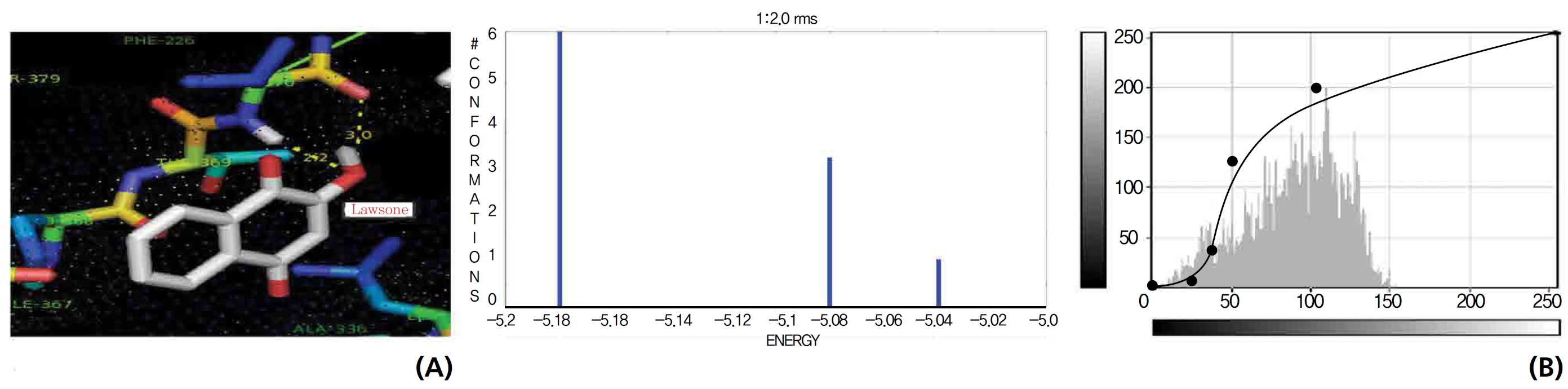
Efficient molecular interactions with G6PDH were studied for lawsone and THN. The results indicate that lawsone shows a better affinity for the docking protein G6PDH than THN does (Table 1). The active sites of one-oxygen and 2-hydroxy (= OH) of the lawsone molecule interacted with oxygen affinity sites of threonine and aspartine at 2.7, 3.4 and 3.6 kcal/mol-1 (Fig. 12). However, the 1, 2, and 3 hydroxy positions of THN, because of the oxygen affinities, turned out to have better amino acid residual interactions with aspartine (2.7 and 2.9 kcal/mol-1), glycine (2.6 kcal/ mol-1), and threonine (2.7 kcal/mol-1) (Fig. 13).
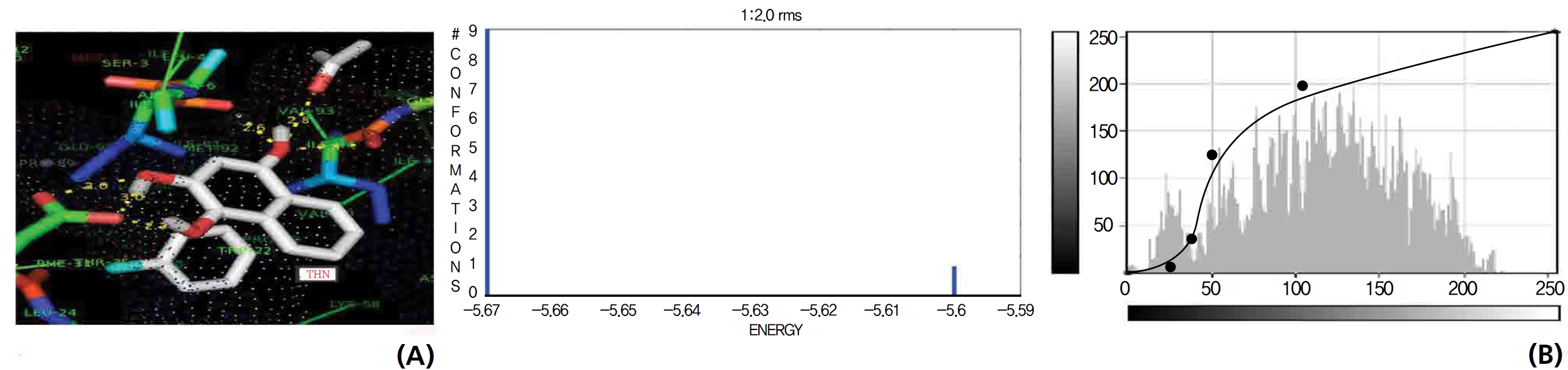
Figs. 14,15 illustrate the interactions of NADPH with lawsone and THN, and the obtained results shows significant molecular affinities. Threonine, alanine, aspartine and leucine of the amino acids made possible interactions with the ortho, the meta and the para positions of lawsone and THN. The macromolecular free force hydrogen-bond interactions of THN were found to have binding affinities of 2.6, 2.7, 2.8, 2.8, 3.0 and 3.0 kcal/mol-1; the lawsone results were 2.9, 3.5 and 3.6 kcal/mol-1 (Figs. 14,15). The results for lawsone binding with threonine and alanine hydrogen bindings through oxygen were found to be 2.9, 3.5 and 3.6 kcal/mol-1.
Since ancient times, medicinal plants, due to their bioactive phytochemicals, have used by humans as treatments for many diseases [7]. Medicinal plants represent complex mixtures of chemical components with biological activities when used concomitantly [8-10]. However, studies have demonstrated that even plants that are used popularly for therapeutic purposes can exhibit hepatotoxicity and nephrotoxicity effects [11-13]. This shows the importance of conducting toxicological studies based on protocols elaborated by regulatory agencies to evaluate the safety of medicinal plants.
Lipophilicity and solubility are the key molecular properties in the absorption of drugs in a biological system. Used to predict the oral bioavailability of a drug, Lipinski’s rule of five (log P, molecular weight, number of atoms, number of hydrogen donors, number of hydrogen acceptors, number of violations, number of rotatable bonds) is considered in the drug discovery process. Computational application tools facilitate learning about the pharmacokinetic properties of individual drug molecules. Many changes and improvements have marked the area of medicinal chemistry, and new approaches to improve drug efficacy by use of technological resources have been developed. In Molinspiration, analyses of the different approaches by using computers are directed to the evaluation of chemistry and physical chemistry data that influences the pharmacokinetic properties of drugs. The molecular and bioactive properties of lawsone and THN are presented in Figs. 16,17 [14]. The selected ligands satisfy Lipinski’s rule of five as log p < 5, molecular weight < 500 g/mol, number of hydrogen bond acceptors < 10, number of hydrogen bond donors < 5 and number of rotatable bonds < 10.
Protein and substrate associations allow us to predict and grade the structures on the basis of their bonds and interactions [15]. Optimization of the synchronous geometrics of the ligands, followed by the docking procedure, enhanced the docking precision and the binding energy [16]. A drug molecule is triggered when the binding of a small molecule to the receptor protein is perfect. Such a protein ligand interaction is comparable to the lock and key principle, in which the lock encodes the protein and the key, is an ensemble by the ligand. The major driving force for forming a bond appears to be a hydrophobic interaction whose specificity is, however, controlled by hydrogen bonding interactions [17]. The receptors remain rigid throughout docking while the ligands are allowed to rotate freely at the active site to achieve site suitable conformations. AutoDock 4.0 generated 10 docking simulations at the active sites of the ligands, and among those, the most stable orientation was chosen according to the value of the binding energy. The most stable conformation possessed minimum energy which explained the mechanism through which the ligand occupied the active site.
Exposure to free radicals from a variety of sources has led organisms to develop a series of defense mechanisms [18]. A fraction of this oxidant inevitably escapes cellular defense, however, and damage to DNA occurs through any of a large complex group of reactions ranging from oxidation of deoxyribose and base moieties to strand breaks. Enzymatic and non enzymatic antioxidants are involved in defense mechanisms such as preventive mechanisms, repair mechanisms, physical defenses and antioxidant defenses. Several of the oxidases are capable of combining oxygen with hydrogen ions derived from different intracellular chemicals to form H2O2. H2O2 is an excited oxygen species that arises during irradiation or as a byproduct of aerobic metabolism and is used in association with CAT, another oxidase enzyme present in large quantities in peroxisomes, to oxidize many substances that might otherwise be poisonous to the cell. The present study concludes that lawsone and THN do not show interactions with H2O2.
Nitric oxide (NO) is a gaseous signaling molecule that readily diffuses across cell membranes and regulates a wide range of physiological and pathophysiological processes, including cardiovascular, inflammation, immune, and neuronal functions. NO is synthesized from L-arginine and molecular oxygen by using calcium calmodulin sensitive NOS. NO has a significant effect on vascular smooth muscle tone and blood pressure through endothelium dependent vasodilation. NO may have an additional role in relaxing the airway’s smooth muscle, thus acting as a bronchodilator. Vascular plaque formation in hypercholesterolemia leads to reduced NO formation; in addition, NO may act as an antioxidant, blocking the oxidation of low density lipoproteins (LDL) and thus preventing the formation of foam cells in the vascular wall. Abnormal activation of platelets is associated with increased platelet adhesion and aggregation and, therefore, a higher incidence of thrombotic events. Platelet activation also leads to the release of smooth muscle mitogens such as growth factors and thromboxane. NO is a potent inhibitor of platelet adhesion and aggregation. Thus, endothelial dysfunction may decrease in NO generation, which leads to abnormal platelet function. Ischemic and reperfusion injuries at the time of organ harvesting, preservation and revascularization initiate myointimal proliferation, which is also promoted by the continuous immune response to an allogeneic organ graft. By reducing free radical toxicity under these conditions, NO may act as a cytoprotective agent, inhibiting platelet and neutrophil aggregation and adhesion to the vascular wall. NO modifies neurotransmitter release in different areas of the brain. According to a proposal, the NO thus produced rapidly diffuses to the pre-synaptic nerve terminal where guanyl cyclase is activated to yield current Good Manufacturing Practic (cGMP), thus facilitating the release of transmitters synthesized in post synaptic sites following the opening of Ca2+ channels and the activation of NOS. The selected ligands show NOS receptor binding affinity through hydrogen bond interactions.
Cells have an elaborated defense system to destroy reactive oxygen species, including CAT (enzymatic) and GSH (non enzymatic), that convert reactive oxygen species to harmless products. CAT is a hemoprotein containing four heme groups, which is found in blood, bone marrow, mucous membrane, the kidneys and the liver. In addition to possessing peroxidase activity, it is able to use one molecule of hydrogen peroxidase as a substrate electron donor and another molecule of H2O2 as an oxidant or electron acceptor. Its function is assumed to be the conversion of the H2O2 produced by
GSH is an essential component to the body’s natural defense system and is composed three amino acids (cysteine, glycine and glutamate). GSH is a primary source of protection for the skin and for the lens, cornea and retina of the eye against radiation damage. It also promotes biochemical detoxification by liver, kidneys, lungs, epithelium of the intestine and other organs through cytochrome P450 activation in the biological system. The ongoing mechanism supports that CAT, GSH receptors, interacted significantly with the ligands of lawsone and THN.
G6PDH is the rate limiting enzyme in the pentose phosphate pathways and produce ribose 5-phosphate and NADH (the intracellular reducing agent in the antioxidant action of peroxidase). A component of the antioxidant system is G6PDH, which is very important for cell survival. G6PDH is the major source of NADPH (the pentose phosphate pathway) and it is used by GSH, thioredoxin systems, to generate reduced forms that will then be used in antioxidant roles. NADPH is a key hydrogen donor for the reduction of oxidized GSH to a tripeptide known as reduced GSH. This tripeptide is used as a reducing agent by GSH peroxidase, which is involved in the detoxification of H2O2. Decreases in the G6PDH activity and as a result, the NADPH level will damage endothelial cells, the kidneys, the liver and red blood cells, which leads to oxidative damage, cellular dysfunction and organ damages. During detoxification of free radicals, GSH is converted to an oxidized form, leading to a reduction in the GSH levels in cellular components. The regeneration of GSH occurs by the action of GSH reductase, which catalyzes the reduction of oxidized GSH to GSH. Of the results for lawsone and THN confirmed molecular interactions with G6PDH and NADPH.
The results of the present investigation are a clear indication that both lawsone and THN can be considered to efficiently bond with NOS, CAT, GSH, G6PDH and NADPH, which was confirmed through hydrogen bond affinities for the respective amino acids. However, H2O2 cannot be consider as having sites to which lawsone and THN can efficiently bond.
The examination of napthoquinone derivatives of lawsone which can be metabolize into 1,2,4-trihydroxy naphthalene by a catalast DTD. In previous experimental results showed the napthoquinone derivatives are ability to induce oxidative damages as consequence of their ability to undergo redox cycling and generation of reactive oxygen species, this phenomenon is considered to be of non toxicological concern of lawsone and its metabolite 1,2,4-trihydroxynapththalene. Lawsone cannot generate reactive oxygen species at a rate sufficient to exceed the capacity of the cells to remove them, and hence no toxicity results. In support of previous study was proved that lawsone itself almost does not show the involvement of hydrogen peroxide generations as it is non redox cycling compound because of stabilizing influence of its 2-hydroxy group. This observation has led to the hypothesis that lawsone and 1,2,4-trihydroxy naphthalene were identical.