Graphene is a two-dimensional, one-atom-thick honeycomb lattice sheet of sp2 carbon atoms. Since the discovery and subsequent development of graphene [1,2], tremendous research has been focused on it due to its large theoretical specific-surface area, strong mechanical properties, excellent thermal and electrical conductivity, and relatively low density [3]. Furthermore, self-assembly is a promising strategy for fabricating graphene-based carbon materials for sensors and catalysis, as well as for energy storage and conversion. Two dimensional (2D) self-assembly of functionalized graphene has been accomplished by means of flow-directed self-assembly [4], evaporation-induced self-assembly [5], the Langmuir-Blodgett technique [6], and layer-by-layer deposition [7]. Recently, 3D carbon aerogels have been obtained by simple assembly of graphene oxide (GO) sheets [8,9]. Compared to traditional carbon aerogels, graphene-based aerogels exhibit ultralow density, high surface area and a very highly porous structure. Typical graphene/carbon composite aerogels were obtained by pyrolysis at 900℃ for 2 h after typical synthesis of polymer solution at 180℃ for 24 h and freeze-drying of the resulting hydrogels [8,9]. Making such freeze-dried, pyrolyzed products is time-consuming and requires an expensive process. Of unusual interest is a report of a process using electron beam irradiation (EBI) techniques that provided products that were nontoxic (without post-processing), used an environmentally friendly procedure, and exhibited the combined effects of cross-linking and sterilization [10]. Moreover, the EBI technique is also useful for modifying the physico-chemical properties of carbon materials. For instance, Park et al. [11] reported that the EBI exposures (50 kGy) resulted in the D to G band intensity ratio of 1.01 (ID/IG) due to the transformation of graphene from the nanocrystalline to the amorphous form. Finally, although reports on poly(vinyl alcohol)-blended hydrogels are numerous, to date, there are only a few reports on wasted-wool-based hydrogels.
Here we report the preparation of self-assembled, wool-based graphene hydrogels as a 3D structure for graphene-based materials. We also report the results of our study of the corresponding mechanical properties, gel fraction, swelling behavior, and hexavalent chromium (Cr(VI)) removal from aqueous solution. We attempted to save time and costs in producing hydrogels using EBI. Properties of self-assembled graphene hydrogels are rarely studied, so this information is of great interest for developing preparations of promising, environmentally friendly, lowcost graphene hydrogels.
Urea, sodium disulfite (Na2S2O5), sodium dodecyl sulfate (SDS), acetone (CH3COCH3), ethanol (C2H5OH), poly(ethylene imine) (PEI, branched, average Mn ~10 000 by gel permeation chromatography, average Mw ~25 000 g/mol by light scattering), PVA (Mw = 85 000-124 000 g/mol, 87-89% hydrolyzed) and potassium dichromate (K2Cr2O7) were purchased from the Sigma Aldrich, Co. (St. Louis, MO, USA). Wasted wool (Merino) was obtained from local bedding factories for free. Disposable sterilized polystyrene square dishes (125 × 125 × 20 mm) were purchased from SPL Life Sciences Co., Republic of Korea.
2.2. Keratin extraction method (S-sulfo keratin)
The wool was vigorously washed with water containing 0.5% SDS, rinsed and then air-dried. Keratin was extracted by sulfitolysis [12,13]. The wasted wool was then extracted using a Soxhlet apparatus and the dried-wool samples (150 g each) were cut into snippets of a few millimeters each, and then placed into 1.5 liters of an aqueous solution containing 8 M urea, 75 g of SDS and 150 g of Na2S2O5. The mixture was heated to 100℃, shaken for 30 min and then filtered through a stainless-steel mesh with pore sizes of 75 μm. The filtrate (molecular-weight cutoff of 12 000 Da) was dialyzed using 15 liters of water that contained 0.1 wt% Na2S2O5, using cellulose tubing, for 3 days at 25℃.
GO was synthesized by a modified Hummers method [14]. Four grams of graphite flakes were added to a 250 mL flask containing 120 mL of H2SO4, which was then stirred for 1 h. Fifteen grams of KMnO4 was gradually added to the mixture at intervals of 20 min (nine times). The mixture was slowly heated to 40℃, and then maintained for 5 h in order to oxidize the graphite. Next, 150 mL of deionized (DI) water was added to the system. Finally, 15 mL of H2O2 solution was added to the mixture, stirred for 30 min, and then left in this condition for 24 h, after which it was centrifuged.
2.4. Preparation of wool-based graphene hydrogels
Hydrogels that contained the prepared keratin protein (5 wt%), PVA (5 wt%), GO (3 wt%), and DI water were produced in the form of a sheet (125 × 125 × 5 mm). To improve the gelation, 0.01 wt% PEI was included in the wool-PVAGO solution. Afterwards, the polymer solution was poured into the square dishes, and the dishes were irradiated with an electron-beam at a dose of 10-60 kGy. The irradiation of the samples was performed using an electron beam accelerator (beam energy 2.5 MeV, beam current 8.5 mA, irradiation width 110 cm, conveyor velocity 10 m/min, dose rate 6.67 kGy/s, and roller type handling system, EBTECH Co., Ltd., Korea) at room temperature, in air. The specific energy (SE) requirement in kilojoules per kilogram (kJ/kg) was equal to the dose in kilogray, as follows:
The tensile-strength-at-rupture and elongation-at-break were measured using a universal test machine (Lloyd, US/LRIOK) according to the ASTM D882 standard method. The data were transferred to a computer to evaluate the stress-strain curve. The gel strength was calculated using Eq. (2) [13].
The sample was dried at 60℃ for 48 h to reach a constant weight (A) and then boiled in double-distilled water for 5 min before being dried again to a constant weight (B). The % gel fraction was calculated using Eq. (3).
The sample was weighed (w1), and placed in distilled water for 72 h, weighed (w2), dried, and weighed (w3) again. The % water uptake was calculated using Eq. (4).
2.8. Cr(VI) adsorption experiments
K2Cr2O7 was used as the source of Cr(VI). Hydrogel (300 mg) was put into a vial containing 30 mL 200 ppm Cr(VI) solution, varying the pH for 12 h. Adjustment of the pH of the Cr(VI) solutions was done using 0.2 M HCl or 0.2 M NaOH. The removal percentage of Cr(VI) was calculated using Eq. (5), where C0 is the initial concentration of Cr(VI) in solution (mg/L), and Ce is the equilibrium concentration (mg/L).
The kinetic adsorption was determined by taking out 0.1- mL samples of the Cr(VI) solution at different times, for each of which the adsorption capacity was calculated using Eq. (6); where Qt is the adsorption capacity at time t, C0 is the initial concentration of Cr(VI) in solution (mg/L), Ct is the concentration at time t (mg/L), m is mass of adsorbent (g), and V is the volume of solution (L). All experiments were performed at 25℃.
The changes in the functional groups of the hydrogels were evaluated using Fourier-transform infrared spectroscopy (FTIR, Varian 1000 FT-IR Scimitar series; PIKE Technologies, USA) on freeze-dried samples. Freeze-dried hydrogel samples were coated with platinum (ion-sputter, Hitachi E-1010, Japan) under vacuum, and were used to investigate the morphology of the keratin protein hydrogels using a Jeol JSM-5900 scanning electron microscopy (SEM). The crystal structure of the hydrogels was estimated using X-ray diffraction (XRD, Rigaku D/ max-2500) with Cu Kα radiation (40 kV, 250 mA). The removal of Cr(VI) was performed by atomic absorption spectrometry (AAS, Perkin-Elmer 3110).
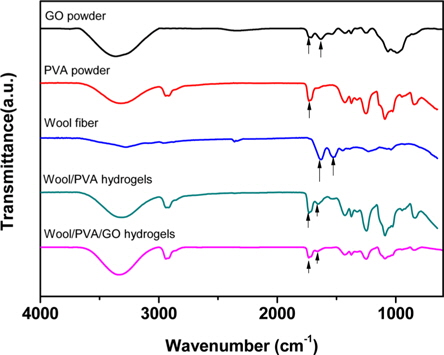
3.1. FT-IR study of the hydrogels
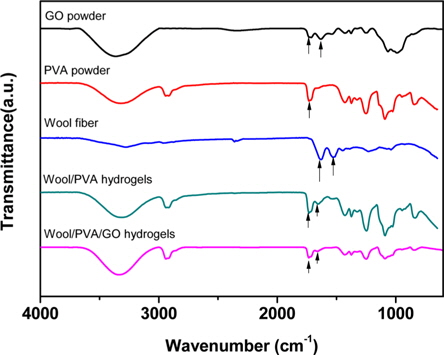
Fig. 1 presents the FT-IR spectra of GO, wool, PVA, wool/ PVA hydrogels and wool/PVA/GO hydrogels. GO has hydroxyl (at 1049 and 3386 cm−1) and ether groups on both sides, with carboxyl groups (at 1736 cm−1) on the edge. At 1624 cm−1, the peak indicated unoxidized graphitic domains in GO, or the stretching vibrations of the C=C bond. Typical, characteristic peaks of keratin proteins were observed, including those indicating the elastic vibration of the C=O bond at 1625 cm−1 and the bending deformation of the C-N-H bond at 1520 cm−1 [15]. A broad band that appeared at 2900-3700 cm−1 belonged to OH groups, and bands of carboxyl and carbonyl groups were also observed at 1736 cm−1 and 1,616 cm−1, respectively [16]. Comparison of wool/PVA and wool/PVA/GO hydrogels, revealed that the locations of peaks for C–H stretching (2920 cm−1), –OH and –NH2 (3700-3000 cm−1), and C=O stretching (1637 cm−1) in NHCO– were similar, and that most of the peaks compared were not significantly different. After adding GO to the polymer solution, the peaks for the oxygen functional groups did not increase and the peak at 1414 cm−1, corresponding to carboxy C–O, was almost eliminated by the EBI treatment. Furthermore, the peak at 1736 cm−1 for the wool/PVA/GO hydrogels decreased. These findings suggested that the functional groups of GO reacted with protein chains and played an important role in reducing GO to graphene when subjected to EBI.
3.2. Characterization of the hydrogels
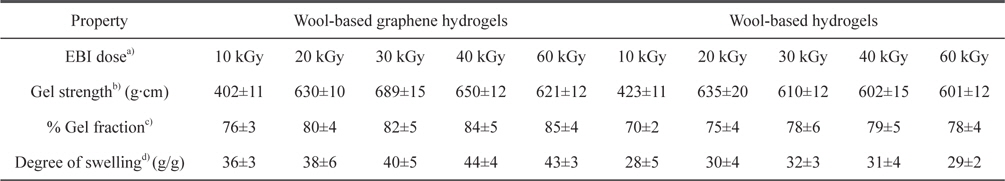
The mechanical properties, gel fraction, and degree-of-swelling of the wool-based graphene hydrogels, and wool-based hydrogels, are shown in Table 1. The mechanical properties and water-absorption ratio of the hydrogels are normally influenced by the degree of cross-linking. The wool-based graphene hydrogels exhibited higher gel strength compared to wool-based hydrogels, when subjected to EBI doses of 20 kGy, with the highest gel strength of 689 g·cm observed at an EBI dose of 30 kGy. The gel strength of the wool/PVA/ GO hydrogels decreased as the EBI dose was increased from 30 to 60 kGy. The gel strength of the wool/PVA hydrogels decreased starting at a lower EBI dose (20 to 60 kGy). In our previous work [13], we observed that keratin-based/PVA hydrogels prepared using EBI exhibited a superior degree of swelling behavior, not only due to the considerable number of hydrophilic components produced upon irradiation (chemical water absorption), but also due to the highly porous structures (physical water absorption). Covalent cross-linking and physical cross-linking probably occurred coincidentally among the S-sulfo keratin main chains, oxygen groups in the PVA aqueous solution, GO, and amine groups of the PEI. Between the polymer chains, radicals were formed via direct or indirect reactions to the radiation, and to solvent-derived radicals. The gel fraction of the wool/PVA/GO hydrogels increased as the EBI dose was increased to 40 kGy, indicating a constant degree of cross-linking. The wool/PVA hydrogels presented probable predominating scission reactions, over cross-linking reactions, at EBI doses above 40 kGy.
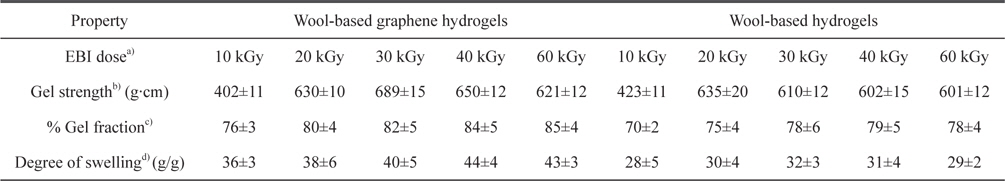
Characterization of wool-based graphene hydrogels and wool-based hydrogels prepared using EBI at various radiation doses
3.3. Surface morphology and structural analysis of hydrogels
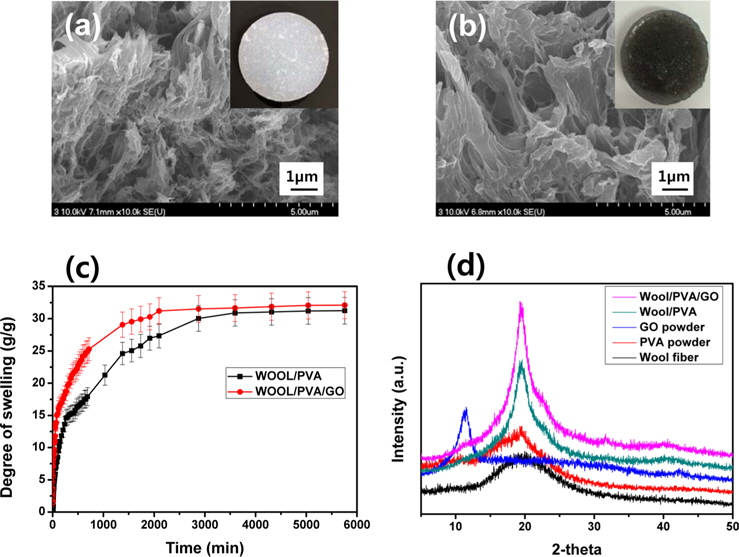
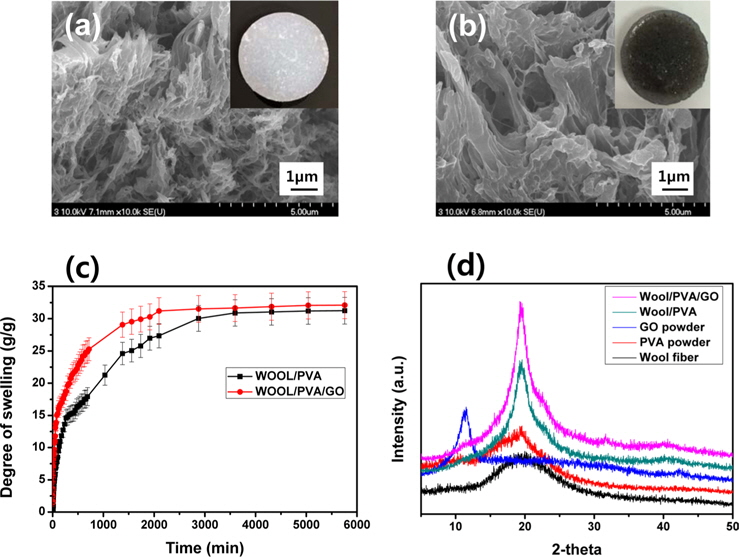
SEM images of the freeze-dried hydrogels produced from wool solutions blended with PVA and GO, in the presence of PEI, and at a dose of 30 kGy; were exhibited in Fig. 2. As seen in Fig. 2a, the freeze-dried wool/PVA/GO hydrogels clearly exhibited a more porous structure than the wool/PVA hydrogels. Furthermore, the wool/PVA/GO hydrogels also exhibited a considerably larger pore size than that of the wool/PVA hydrogels. A number of the physico-chemical properties (e.g., the gel fraction, degree of swelling, gel strength, and swelling kinetics) of the resulting hydrogels can be influenced by pore size [13]. The types and amounts of protein have probably influenced the morphological properties of the resulting hydrogels. Higher swelling capacities and higher swelling rates are required in practical applications. The swelling kinetics in water of wool/ PVA/GO and wool/PVA hydrogels is represented in Fig. 2c. The freeze-dried wool/PVA/GO hydrogels absorbed distilled water rapidly (more than 28 g/g within 2000 min) and then reached a plateau, whereas wool/PVA hydrogels absorbed approximately 25 g/g in the same time. The swelling kinetics of superabsorbent polymers is influenced by the smaller pore size of the absorbents. However, the swelling behavior of the wool/PVA/GO and wool/PVA hydrogels was dominated by larger pore size after 30 min. This finding suggests that wool/PVA/GO hydrogels have the potential for practical applications because of their excellent swelling capacity and swelling kinetics. To further explore the structural changes of the distance of layers, a series of XRD analyses were carried out and the results presented in Fig. 2d. For the GO sample, an intense and sharp diffraction peak shows at 2θ = 10.6°, corresponding to the (001) reflection [17]. Wool has a broad peak at 2θ = 20.2°, and typical PVA at 2θ = 19.5°. The XRD pattern of wool/PVA/GO hydrogels was similar to those of wool/PVA hydrogels, while the peaks characteristic of GO completely disappeared. A broad diffraction peak at around 2θ = 21°, and a very weak peak at 2θ = 44° corresponding to the (100), suggested significant changes in the crystal structure of GO and confirmed again the reduction of GO. This is in accordance with the above FT-IR results for self-assembled, woolbased graphene hydrogels.
3.4. Removal of Cr(VI) by self-assembled, wool-based graphene hydrogels
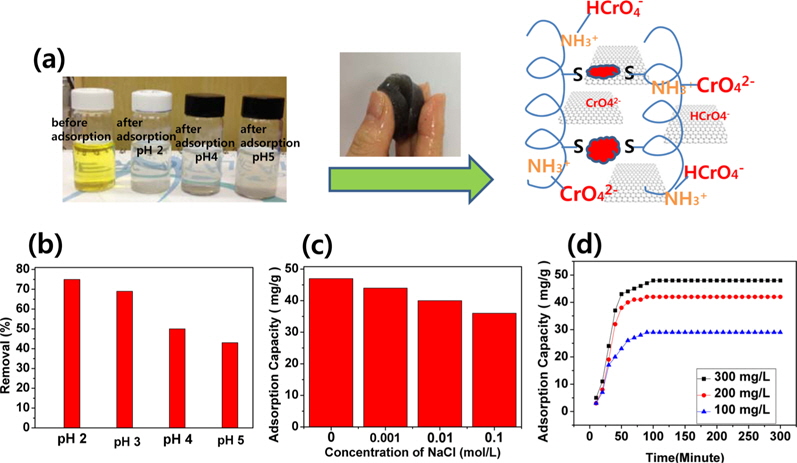
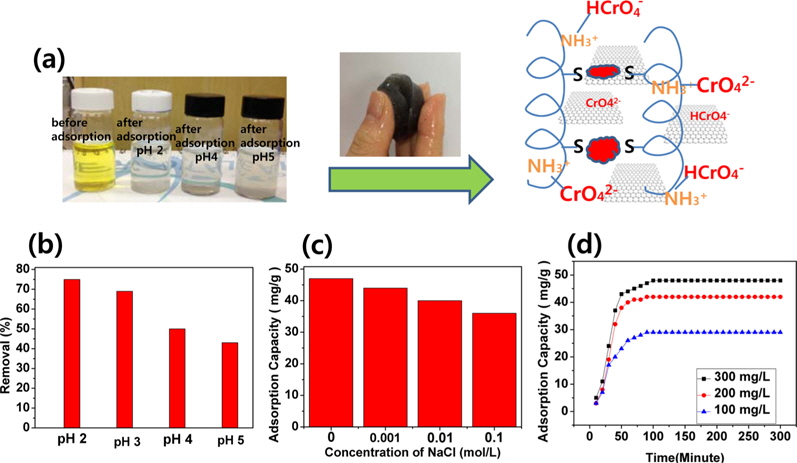
A hypothetical schematic is presented in Fig. 3a. Wool comprises a three-dimensional mesh structure of keratin, which is composed of polypeptides with different amino acids, with inter- molecular bonding by disulfide cystine amino acids. Sulfur side-chains on the cysteine molecules after sulfitolysis of wool were bonded to another cysteine by the EBI. A slight shift of the typical absorption peaks for the C=O and C-N-H bonds showed in the FT-IR spectra of the self-assembled, wool-based graphene hydrogels, due to intermolecular interactions and cross-linking reactions [13]. In Fig. 3b, the effect of pH on the adsorption of Cr(VI) is significant, and is possibly due to the amine groups in self-assembled, wool-based graphene hydrogels [18]. At pH 2.0-6.0, the predominant Cr(VI) species initially exists in the monovalent (HCrO4 −) form; then is gradually converted to the divalent (CrO4 2−) form by the increased pH. As the pH increased, the uptake of Cr(VI) ions decreased because the higher concentration of OH- ions present in the aqueous solution competed with the Cr(VI) species (CrO4 2−) for the adsorption sites [18]. By adjusting the initial pH to 2.0 before adsorption, and by varying the concentration of NaCl, the effect of ionic strength on the adsorption of Cr(VI) by self-assembled, wool-based graphene hydrogels was studied. The results are presented in Fig. 3c. The adsorption capacity remained approximately 82%, whether with no NaCl or with 0.1 mol/L NaCl. That is to say, the effect of ionic strength was not very great. The Cr(VI) adsorption kinetics, of hydrogels at pH 2.0, are shown in Fig. 3d. The adsorption capacity of Cr(VI) increased according to the adsorption time and concentrations of Cr(VI) solution. The equilibrium capacity was 29, 41, and 49 mg/g for 100, 200, and 300 mg/L of initial Cr(VI) solution, respectively. After 12 h, the color of the Cr(VI) aqueous solution changed from yellow to colorless.
Here, we reported the preparation of self-assembled, woolbased graphene hydrogels to save time and costs in producing hydrogels. Promising, environmentally friendly, low-cost graphene hydrogels were successfully prepared by means of EBI and then used as an adsorbent for Cr(VI) ions from aqueous solution. The resulting self-assembled graphene hydrogels exhibit fibrous and highly porous morphologies and graphene structures, qualities confirmed by SEM and XRD analyses. These findings are in good agreement with the results of the FT-IR analysis. Such an EBI-induced self-assembly process for producing graphene offers a more convenient, more environmentally friendly and less expensive technique for large-scale applications.