활성탄은 대단히 넓은 비표면적을 가지는 탁월한 흡착매로서 정수, 석유화학제품 분리 및 정제, 의약품 정제, 그리고 유독성 유기물질, 냄새 및 색도유발물질, 금속이온 등을 흡착제거 하는 데 사용되어 왔다. 또한 활성탄은 촉매 및 촉매 담체, 가스저장매체 등으로도 사용되고 있다[1,2]. 현재 사용되고 있는 활성탄은 입자 크기와 형태에 따라 분말, 입상, 구형, 그리고 펠릿 활성탄으로 구별한다. 또한 활성탄의 물성은 비표면적(specific surface area), 비용적(specific pore volume), 요오드가 등으로 나타낸다.
활성탄 제조에 사용되는 원료물질은 높은 탄소함유율, 최소무기물질 함유량, 이용 가능한 양과 가격, 저장기간, 작업성 등을 고려하여 선택된다. 원료물질의 선택은 제조공정조건과 더불어 제품 활성탄의 물성을 좌우하는 가장 중요한 역할을 한다. 현재 주로 사용되고 있는 원료로는 나무, 석탄, 야자껍질 등이 있다. 이 중 국내에서는 야자껍질이 가장 많이 사용되고 있다.
최근에는 고분자물질을 원료로 활성탄을 제조하는 연구가 활발하게 이루어지고 있다. 이렇게 제조되는 활성탄은 사용되는 고분자의 선택에 따라 다양한 물리적, 화학적 특성을 갖도록 제조될 수 있다[2-4].
이온교환은 고체 이온교환수지 입자와 액체사이에 이온들의 가역적이고 양론적인 교환이다. 이온교환기술은 고순도 물의 제조뿐만 아니라 연수화, 해수담수화, 폐수처리 등과 같은 일반적인 수처리 분야에 널리 사용되고 있고, 화학공장에서 화합물 정제에 사용되는 분리공정의 한 가지 방법이다. 이때 사용되는 이온교환수지(ion exchange resin)는 주로 구형의 알갱이 형태가 사용되는데 교환하는 이온에 따라 양이온교환수지와 음이온교환수지로 나뉘고, 그 화학적 성질에 따라 강산성, 약산성, 강염기성, 약염기성 등으로 구별한다. 실제 공정에서 이온교환수지의 교환용량이 포화에 도달하여 허용 한계농도 이상으로 해당 이온들의 유출이 시작되면, 처리공정은 이온교환수지의 재생공정으로 전환되고, 적절한 재생공정이 종료되면 다시 처리공정으로 전환된다. 이렇게 이온교환수지는 주기적인 재생공정을 통하여 장기간 사용될 수 있다. 그렇지만 이온교환수지의 사용기간이 길어짐에 따라 즉, 재생횟수가 증가함에 따라 이온교환성능은 점차 저하된다. 어느 정도 이상으로 교환성능이 저하된 이온교환수지는 더 이상 사용될 수 없으므로 폐기되고 새로운 수지가 사용되게 된다. 이때 발생되는 폐이온교환수지(waste ion exchange resins)는 다양한 응용분야에 따라 상당한 양이 생성될 것으로 예상된다[5,6].
폐이온교환수지는 처리되어야 하는 고형폐기물로서 환경적 측면뿐만 아니라 처리비용에 따른 경제적 측면이 고려된다. 따라서 폐이온교환수지의 재활용 방안이 마련된다면 언급한 두 가지 측면이 모두 해결될 수 있을 것이다. 더불어 재활용으로 인한 이익창출 또한 기대할 수 있을 것이다.
정수용으로 주로 사용되는 이온교환수지는 술폰화폴리스티렌(sulfonated polystyrene)과 디비닐벤젠(divinylbenzene, DVB)의 공중합으로 제조되며 활성탄제조의 원료로 사용될 경우 탄화(carbonization)공정과 활성화(activation)공정 내내 구형을 유지하고, 균일한 내부구조와 높은 탄소함유율을 가질 뿐만 아니라 여러 적용분야에 사용되기 적합할 정도의 상당한 기계적 강도를 나타내므로 매우 우수하고 적합한 활성탄 제조 원료물질이라 할 수 있다. 또한 구형의 활성탄은 우수한 수력학적 특성으로 고정층(fixed-bed) 형태의 분리공정에 특히 유용하다[2,7]. 다양한 규격으로 제조된 활성탄은 GC나 HPLC 등과 같은 크로마토그래분야, 천연가스의 저장용 흡착매분야, 일반 정수처리분야, 폐수처리분야 등 다양한 분야에 활용될 수 있는 것으로 보고되고 있다[4,8,9]. 또한 탄화공정에 기존의 활성탄 제조 및 재생설비를 활용할 수 있을 것으로 기대되어, 제조기술의 개발 및 상용화는 그 경제적 효과가 탁월할 것으로 기대된다.
본 연구에서는 이온교환수지를 탄소원으로 하여 탄화와 활성화 공정을 통하여 활성탄의 제조 시 다양한 물성의 활성탄을 제조하는데 공정변수의 영향이 실험적으로 연구되었다. 활성탄의 물성으로 비표면적과 비용적이 사용되었고, 이를 제어하는 공정변수로 탄화시간 및 온도, 활성화시간 및 온도, 승온속도가 선정되었다. 또한 폐이온교환수지에 적용을 고려하여 이온교환수지에 이미 흡착된 이온이 제조된 활성탄에 미치는 영향이 연구되었다.
활성탄은 탄화공정과 활성화공정의 두 가지 주요 단위공정을 통하여 제조된다. 탄화공정에서는 원료물질에 포함되어 있는 산소, 수소와 같은 탄소 이외 물질이 열분해로 대부분 제거된다. 이를 위하여 공정은 보통 800 ℃ 정도의 온도에서 불활성가스 분위기에서 이루어진다. 이때 남겨진 탄소는 판상구조를 형성하고, 이러한 판상구조물들은 서로 불규칙적인 배열을 하게 된다. 이러한 불규칙적인 배열로부터 형성된 간극은 활성탄 기공(pore)의 기초가 된다. 생성된 탄화물은 활성화공정을 통하여 활성탄의 높은 비표면적과 특정 기공용적(pore volume)의 활성탄이 만들어진다[1].
활성화공정의 목적은 탄화공정에서 만들어진 간극의 직경을 확장시키고, 기공용적을 증가시키고, 약간의 새로운 기공용적을 창조하고, 결과적으로 비표면적을 증가시키는 것이다. 기공의 구조와 기공크기분포(pore size distribution)는 활성화공정의 방법 및 운전조건뿐만 아니라 원료물질의 특성과 탄화공정조건에 크게 의존한다. 이와 같은 공정의 목적을 달성하기 위하여 활성화공정에는 화학적 활성화(chemical activation)와 물리적 또는 열적 활성화(physical or thermal activation)가 사용된다. 활성화공정의 종류와 공정조건에 따라 제조된 활성탄의 물성 즉, 마이크로세공(micropore), 메소세공(mesopore), 매크로세공(macropore)과와 같은 기공크기분포, 활성탄입자의 강도, 비표면적, 기공용적 등이 변하게 된다[1,3,10,11].
물리적 활성화공정은 증기, 이산화탄소, 이들의 혼합가스와 같은 적절한 산화제의 존재 하에 800~1,100 ℃의 온도에서 수행되는 산화반응이다. 고온에서 이산화탄소 또는 증기로부터 발생하는 활성산소는 활성탄을 구성하는 탄소 중 반응하기 쉬운 부분과 반응하여 아래 반응식과 같이 연소 또는 기화된다.


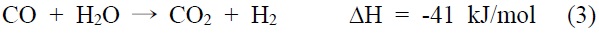
이러한 활성화 반응은 두개의 과정으로 일어난다. 첫 번째 과정은 탄화공정에서 판상구조로 배열되지 못하고 간극에 채워져 있는 탄소가 우선 연소되어 막힌 기공을 뚫어주는 역할을 한다. 두 번째 단계에서 판상으로 배열된 탄소가 연소되어 활성자리(active site)를 생성하고 기공을 넓혀주게 된다. 따라서 물리적 활성화공정에서 제품 활성탄소의 물성에 영향을 주는 인자로는 사용된 산화제 가스의 종류, 활성화온도, 활성화 시간 등이 있다.
본 연구에서는 탄화공정에 질소가 사용되었고, 이산화탄소를 산화제로 사용하여 물리적 활성화공정이 사용되었다.
본 연구에 사용된 이온교환수지는 (주)삼양사의 다공형(porous type)의 강산성 양이온수지 중 수처리에 일반적으로 사용되는 TRILITE CMP 16이 사용되었다. 활성탄의 비표면적과 비용적을 고려하여 겔형(gel type)이 아닌 다공형을 사용하였다. CMP 16의 물리화학적 물성은 Table 1과 같다.
이온교환수지는 탄화공정에서 만들어진 기공 구조와 비표면적은 활성화공정에서 개량되어 제품 활성탄은 다양한 크기의 기공을 가지게 된다. 마이크로세공과 메소세공은 기상이나 액상으로부터 다양한 형태의 화학물질의 대량 흡착에 사용되므로, 활성탄 제조 시 이러한 형태의 기공을 형성시키는 것은 대단히 중요하다. 따라서 적용분야에 적합한 크기의 기공을 갖는 활성탄을 제조하기 위하여 탄화 및 활성화공정 조건에서 기공 크기를 제어하는 데 중요한 인자를 찾고, 그에 대한 영향을 규명하는 것은 대단히 중요하다.
이온교환수지로부터 활성탄제조의 수율을 판단하기 위하여 사용된 이온교환수지의 함수율을 실측하여 Table 1과 비교하였다. 실험은 이온교환수지 10.0 g을 정량하여 비이커에 담은 후 105 ℃ 건조용 오븐에 투입하고 12시간 이상 건조 후 질량변화가 없음을 확인하고 함수율을 실측하였다. 본 연구에서 사용된 CMP 16의 실측된 함수율은 54.9%로 Table 1의 46~52%보다 높았다. 시료의 건조감량과 수율을 계산하는 데 실측치가 사용되었다.
본 연구에서는 다양한 비표면적과 비용적의 활성탄을 제조하는 공정변수로 탄화온도, 활성화온도, 승온속도, 탄화와 활성화 공정에서 유지시간이 선정되었다. 공정변수 각각에 대하여 비표면적과 비용적을 제어할 수 있는 적절한 운전범위를 실험적으로 규명하였다. 또한 폐이온교환수지를 인공적으로 모델화하기 위하여 Ca2+와 Mg2+으로 포화된 수지를 사용하여 활성탄을 제조 비표면적과 비용적을 비교 검토하였다.
실험에 사용된 장치는 관형전기로(tube furnace), 알루미나보트,
[Table 1.] Physicochemical properties of CMP 16[12]
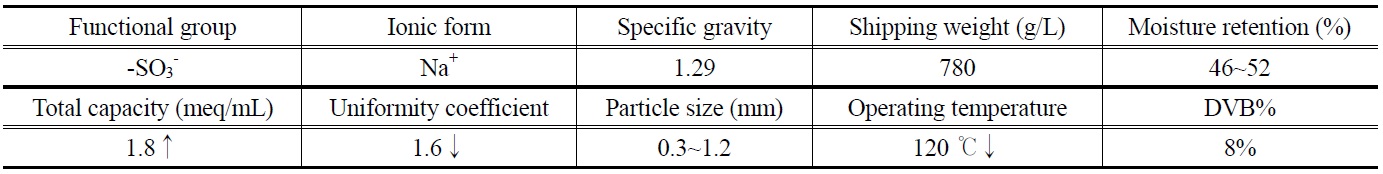
Physicochemical properties of CMP 16[12]
가스실린더, 그리고 가스유량계로 등으로 구성된다. 관형전기로는 내경 2 in의 알루미나관과 석영관, 발열체(SiC), 열전대(thermocouple), 온도제어기, 가스투입부, 가스배출부 등으로 구성되며, 최대운전온도 1,700 ℃ 그리고 최대승온속도 5 ℃가 가능하다. 균일한 온도로 가열이 가능한 구역은 관형전기로 중심 10 cm이다. 운전 시 길이 10 cm의 알루미나보트를 가열부분에 배치하였다. 알루미나관 내부에 미세한 압력을 생성하여 균일한 가스분위기를 만들어 주기 위하여 가스투입부와 배출부에 가스 투입 및 배출부분이 축소된 스테인레스 스틸 뚜껑을 만들어 설치하였다. 탄화와 활성화 시간, 온도, 승온속도 등은 온도제어기로 설정하였다.
관형전기로는 시료를 투입하기 전에 온도제어기에 탄화온도, 승온속도, 탄화온도에서 체류시간, 활성화온도, 승온속도, 활성화온도에서 체류시간 등을 프로그래밍하였다.
이온교환수지는 제조사에서 구입한 그대로 알루미나보트 (길이 100 mm, 폭 20 mm, 높이 13 mm)에 수지 약 6 g을 정량하고 철사로 제작한 투입기구로 조심스럽게 관의 중심 즉, 균일한 온도구간으로 투입한다. 질소가스를 1.0 L/min의 속도로 투입하며 탄화공정을 시작한다. 설정한 승온속도로 가열하여 탄화온도에 도달하면 정해진 탄화시간동안 온도를 유지시킨 후 다시 활성화온도까지 승온시키고 활성화시간동안 온도를 유지시킨다.
활성화시간이 되면 CO2를 1.0 L/min의 속도로 투입하기 시작한다. CO2의 투입은 두 가지 방법으로 수행되었다. 즉, 탄화온도 보다 높은 온도에서 활성화를 시키는 경우 첫 번째 방법은 탄화종료 후 활성화온도로 승온하는 시점에서 CO2를 투입하고, 두 번째 방법은 활성화온도로 승온 중엔 질소로 운전하고 활성화온도 도달시점에서 CO2를 투입하기 시작하였다. 활성화 종료 후 냉각운전 중에는 질소를 계속 공급하였다.
60 ℃ 이하로 냉각된 시료는 투입 때 사용된 동일한 기구로 꺼내어 비닐백에 넣은 후 비표면적과 비용적 분석을 위하여 데시케이터에 보관하였다.
제조된 활성탄 시료는 비표면적 및 비용적 측정장치인 미국 Quantachrome사의 모델 NOVA 4000e로 분석되었다. 비표면적은 BET (Brunauer-Emmett-Teller)법으로, 비용적은 BJH (Barrett-Joyner-Halenda)법으로 측정하였다.
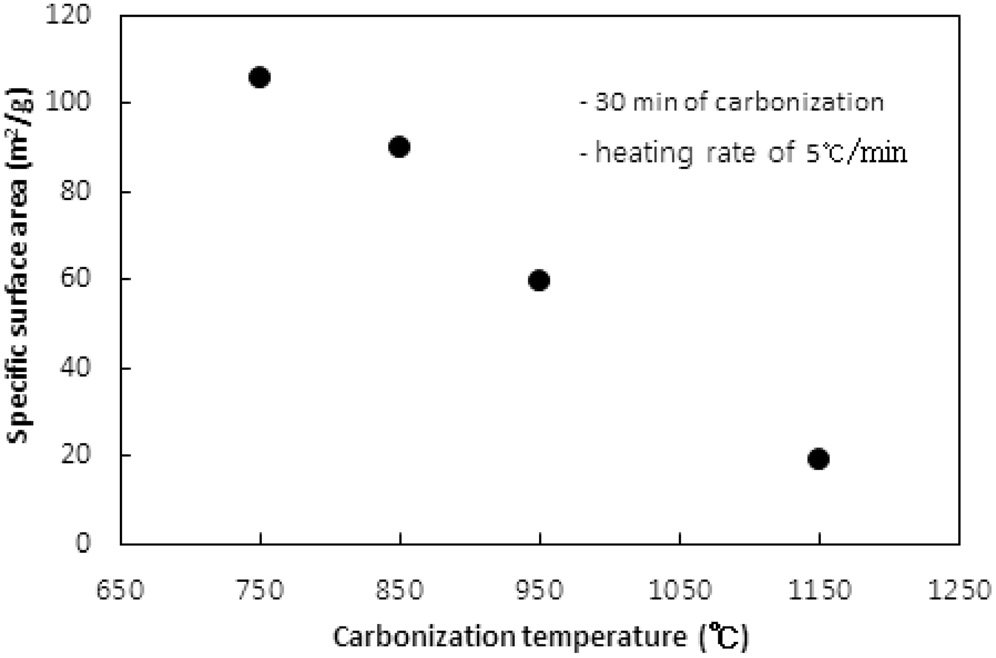
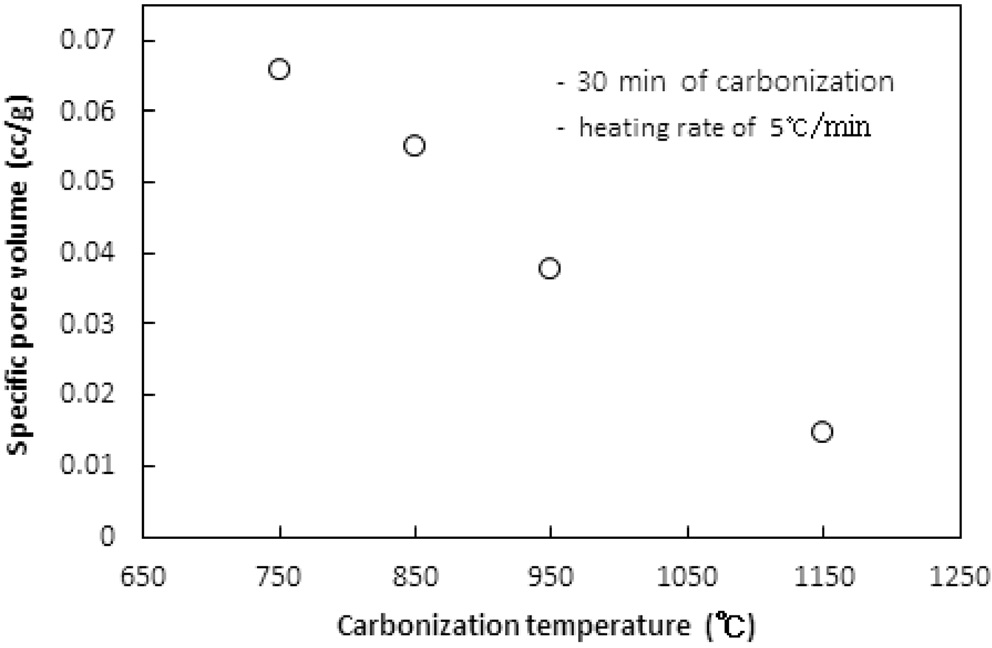
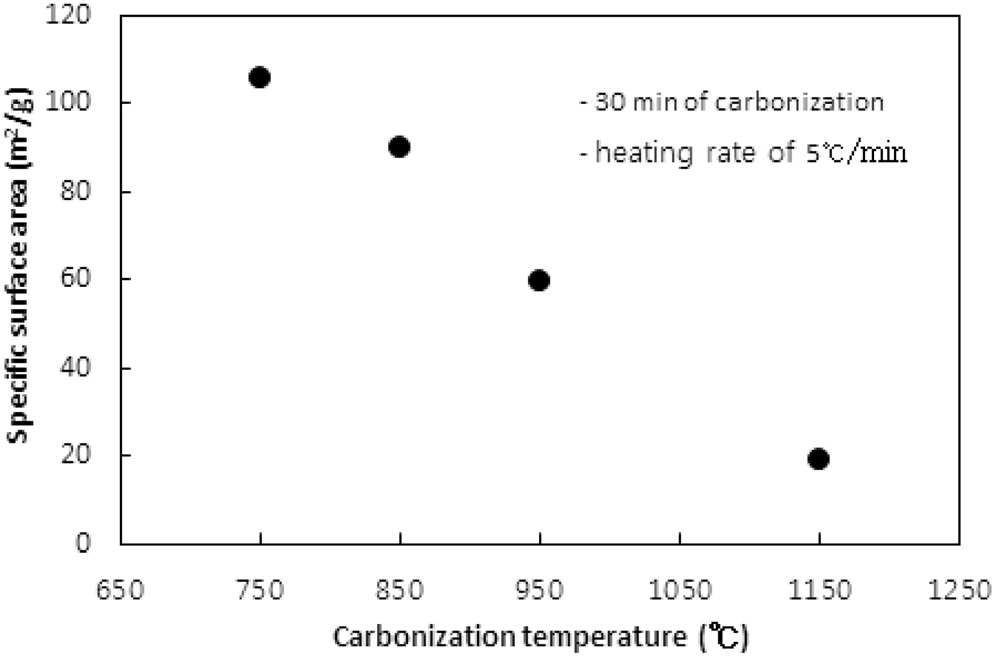
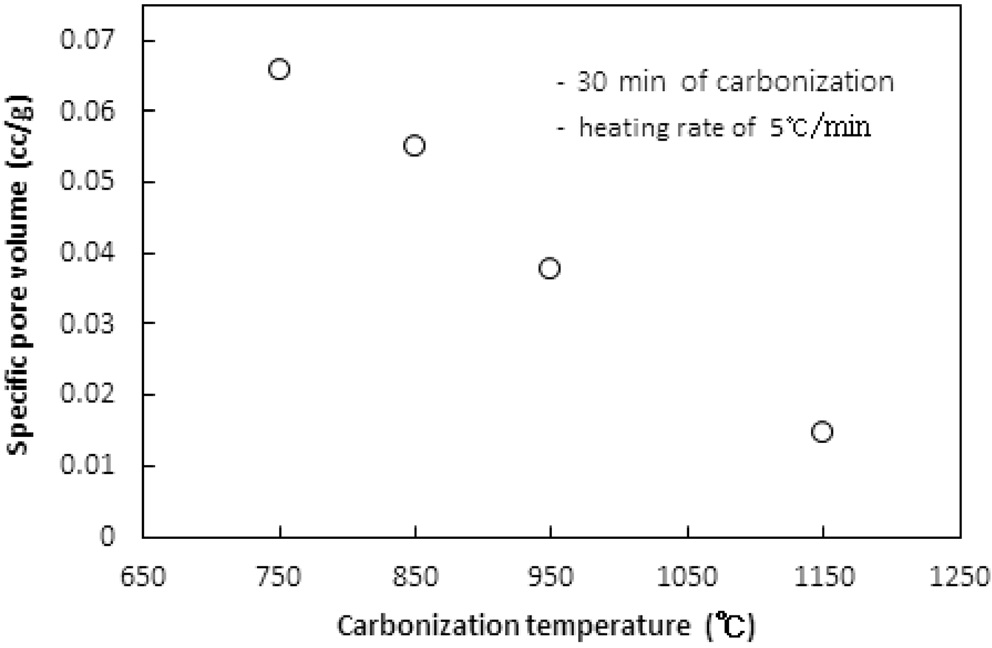
고분자 물질인 이온교환수지를 구성하는 원소 중 탄소를 제외한 산소, 수소 등을 열분해로 대부분 제거해 주는 탄화공정에서 탄화온도에 따른 탄화물의 비표면적과 비용적은 Figure 1과 2와 같다. 실험 시 이온교환수지는 6.0 g이 투입되었고, 활성화 없이 탄화공정만 수행되었다. 탄화온도까지의 승온속도는 5 ℃/min였고, 탄화시간은 30분이었다. 질소가스의 투입속도는 1.0 L/min이었다.
탄화공정은 200 ℃ 이하의 저온에서 흡착 및 자유수분이 방출되고, 300~400 ℃에서 폴리스티렌-DVB 구조의 분해로 해중합(depolymerization)과 술폰 이탈(desulfonation)이 발생하고, 500 ℃ 정도에서 폴리스티렌과 DVB의 분해가 발생하는 것으로 알려진다[10]. 따라서 750 ℃ 이상의 탄화온도는 탄화공정에 충분하다.
탄화온도가 750, 850, 950, 1,150 ℃로 증가함에 따라 비표면적은 106 m2/g에서 18.8 m2/g로, 비용적은 0.0659 cm3/g에서 0.0147 cm3/g로 탄화온도가 상승함에 따라 비표면적과 비용적이 선형적으로 감소하는 경향을 보였다. 이러한 현상은 탄화공정
[Table 2.] Production yield for the change of carbonization temperature

Production yield for the change of carbonization temperature
시 수지에서 열수축(thermal shrinkage)이 발생하여 비표면적과 비용적이 감소하는 것으로 해석된다. 열수축은 탄화공정 시 고온에서 탄소이외의 물질이 분해 제거되며 생성되는 대부분의 초미세공(ultramicropore) 및 마이크로세공과 기존 이온교환수지에 존재하는 기공이 쪼그라들어 소멸되는 현상으로 탄화온도가 증가함에 따라 열수축이 심하게 된다[8]. 열수축이 너무 심해지면 다음 공정인 활성화공정에서 산화제가 침투할 수 있는 간극이 사라져 충분한 비표면적과 비용적을 생성하기 어렵다.
1,000 ℃ 이상의 온도에서 탄화가 수행될 경우 과다한 열수축으로 비용적과 비표면적이 너무 작아져 다음 공정인 활성화공정에서 얻을 수 있는 비표면적과 비용적의 최대치가 낮아질 것으로 사료된다.
Table 2는 탄화 후 시료의 질량과 건조기준으로 수율을 나타낸다. 탄화 후 남은 시료의 질량은 탄화온도에 따라 거의 변화가 없었다. 즉, 탄화공정에서 제거되는 탄소 외 물질의 총량은 화학적으로 일정하므로 질량 감소율은 거의 일정하지만, 탄화온도에 따라 열수축의 정도가 변화하는 것으로 판단된다. 따라서 수율은 평균 44%로 거의 일정하였다.
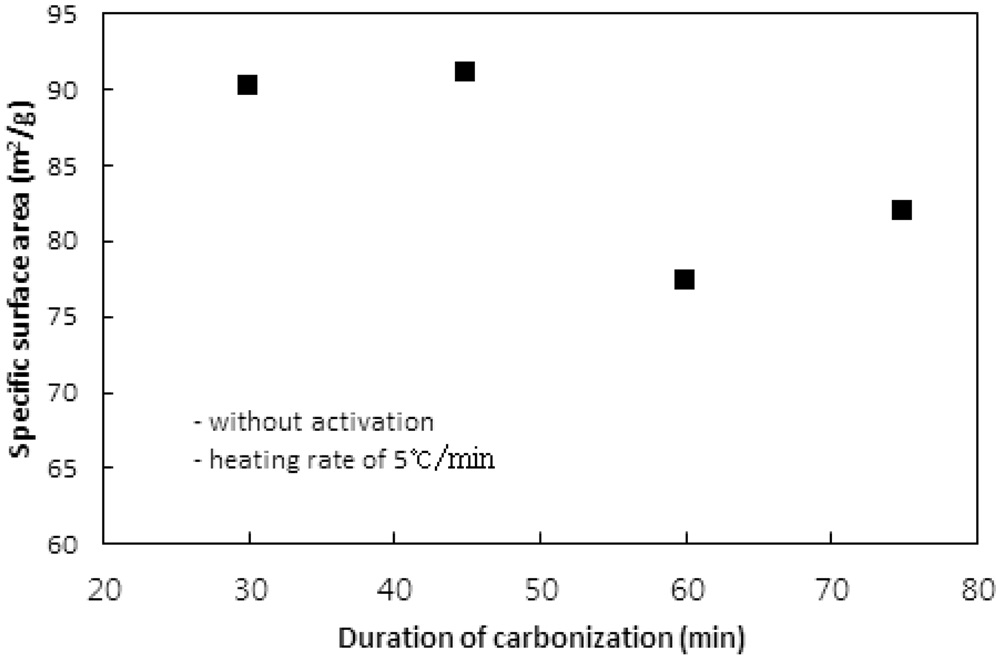
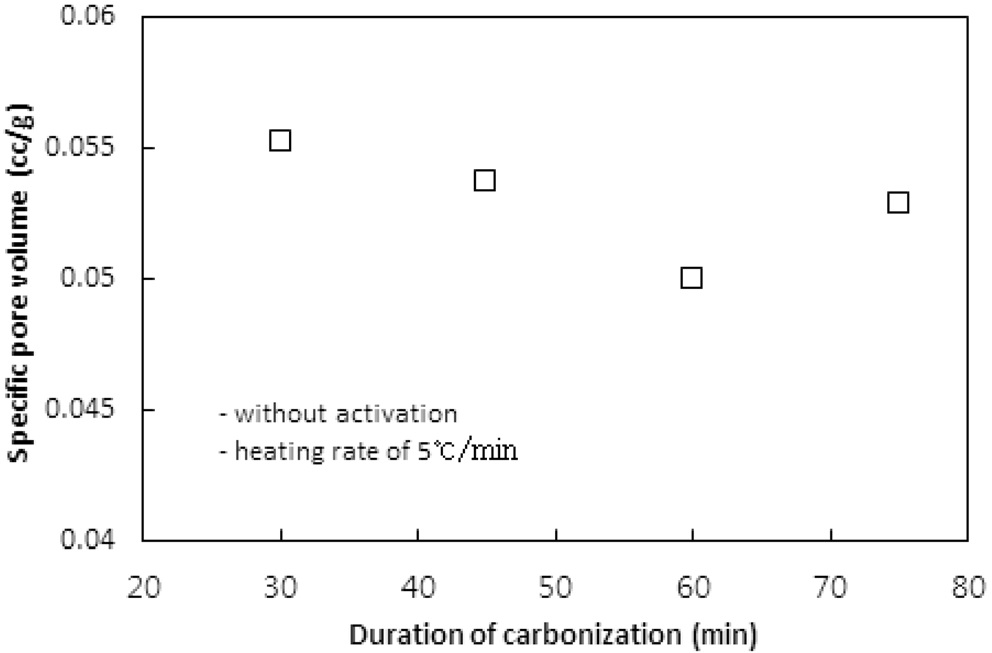
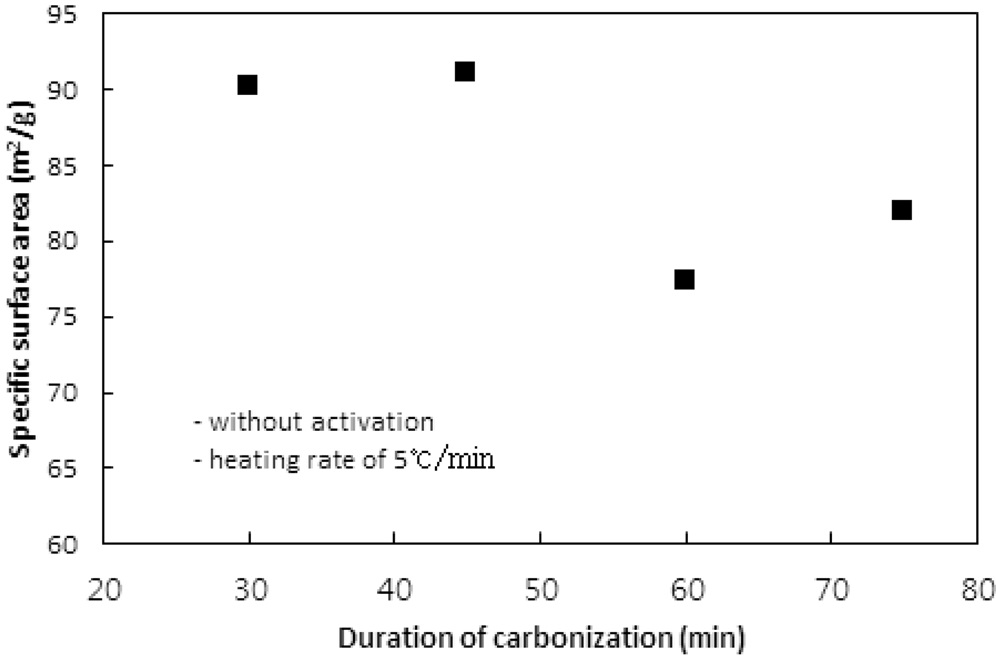
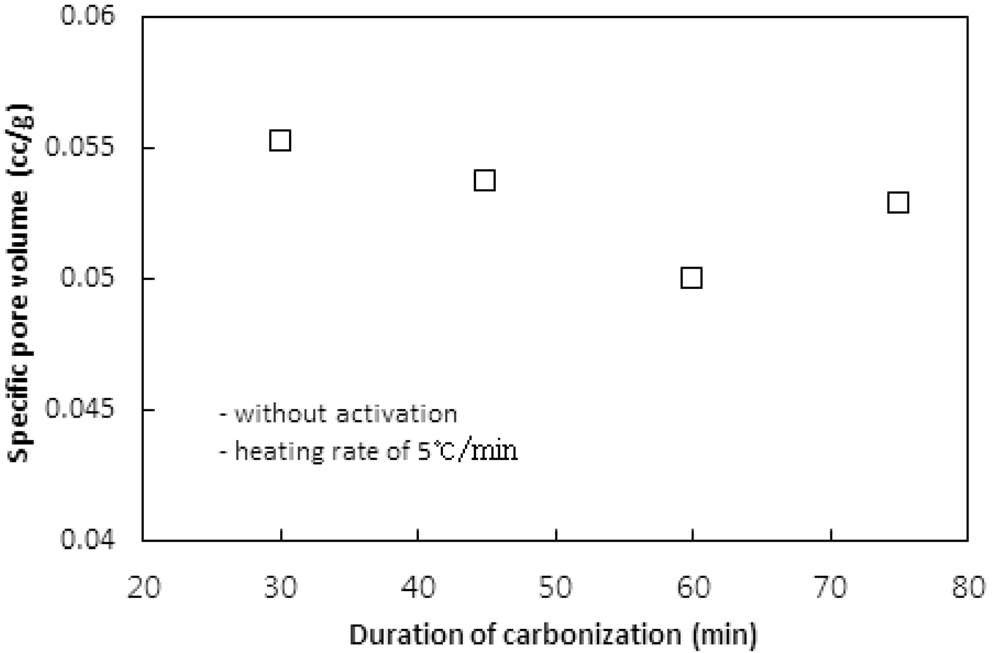
탄화공정에서 탄화시간에 따른 탄화물의 비표면적, 비용적 그리고 수율에 대한 영향을 평가하기 위하여 탄화온도와 승온속도를 850 ℃와 5 ℃/min로 고정시키고 탄화시간을 30, 45, 60, 75분으로 변화시키며 탄화공정만을 수행하였다. 탄화시간에 따른 비표면적과 비용적의 변화는 Figure 3과 4와 같고, Table 3은 수율을 보여준다.
탄화시간이 45분까지는 비표면적에서 큰 차이는 없었지만
[Table 3.] Production yield for the change of carbonization time

Production yield for the change of carbonization time
그 이상의 시간에서는 비표면적이 10% 이상 감소하였다. 이는 탄화공정에서 발생되는 기공은 주로 초미세공이다. 문헌에 따르면 초미세공의 기공크기는 0.7 nm 이하 또는 0.4 nm이하로 정하고 있다[1,10]. 탄화시간이 길어짐에 따라 열수축이 심해져 내부의 초미세공 일부분이 비표면적 측정 시 사용되는 질소분자가 진입할 수 없을 정도로 수축, 소멸되기 때문인 것으로 판단된다. 비용적의 경우도 비표면적의 결과와 같은 경향을 보여준다.
수율의 경우 60분까지는 Table 2에서와 같은 경향으로 평균 44% 정도를 보였지만 75분의 탄화시간에서는 증가하였다. 이는 탄화시간 동안 충분한 분해반응으로 탄소이외의 물질이 더 많이 제거되기 때문인 것으로 사료된다. 이러한 결과는 Nakagawa et al.[13]이 감량을 이온교환수지를 구성하는 폴리스티렌체인의 분해로 발생되는 벤젠과 톨루엔 같은 방향족화합물의 생성에 기인하는 것으로 해석한 것에 상응한다. 탄소의 순도 측면에서 긴 탄화시간이 선호될 수 있으나 비표면적과 비용적 측면에서는 30분이면 충분한 탄화가 일어난 것으로 판단할 수 있다.
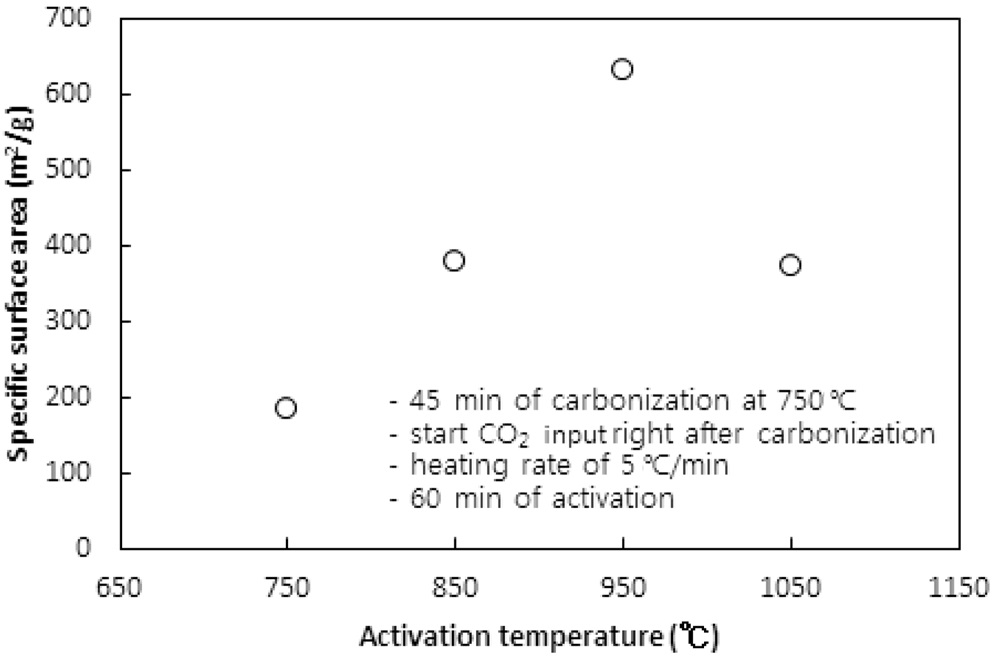
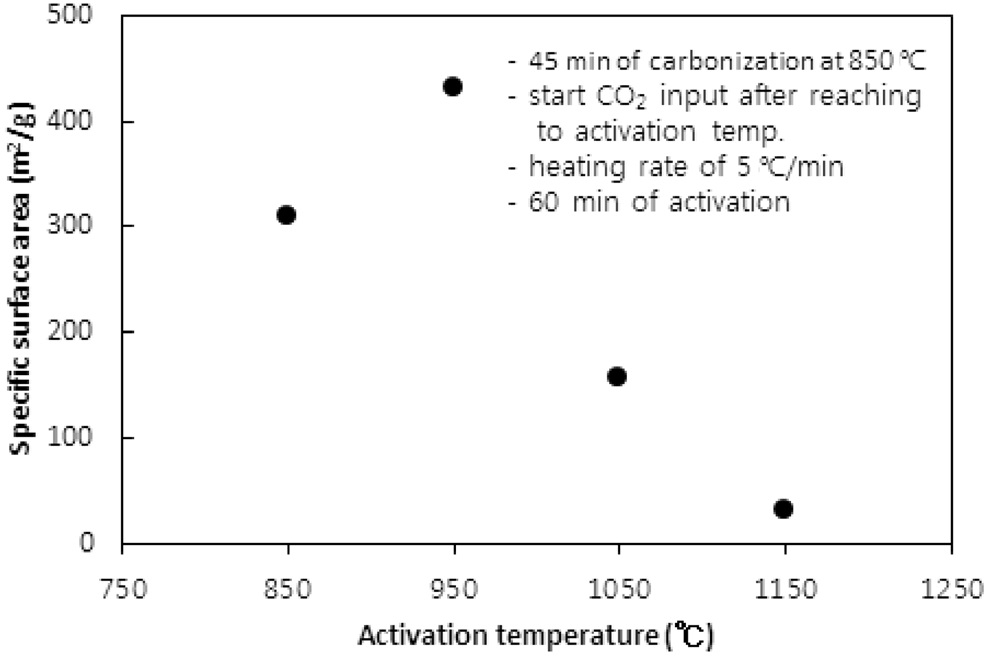
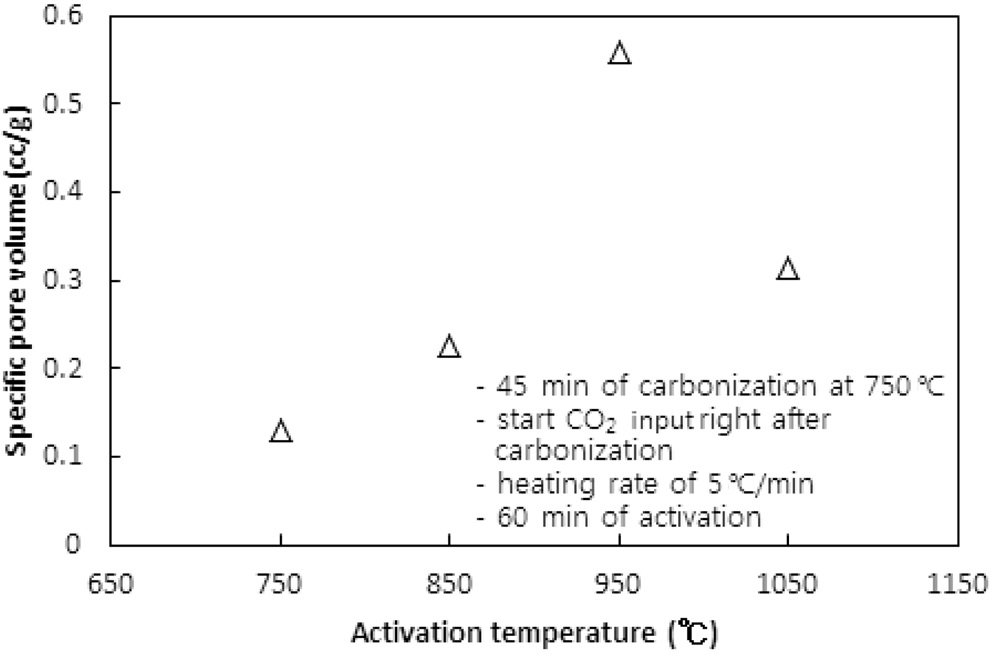
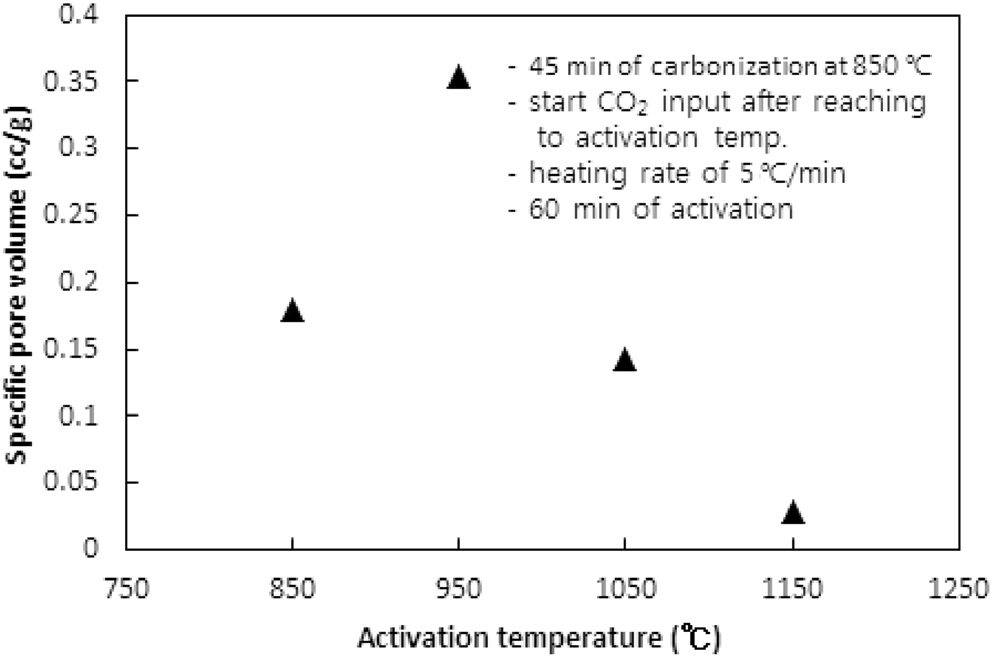
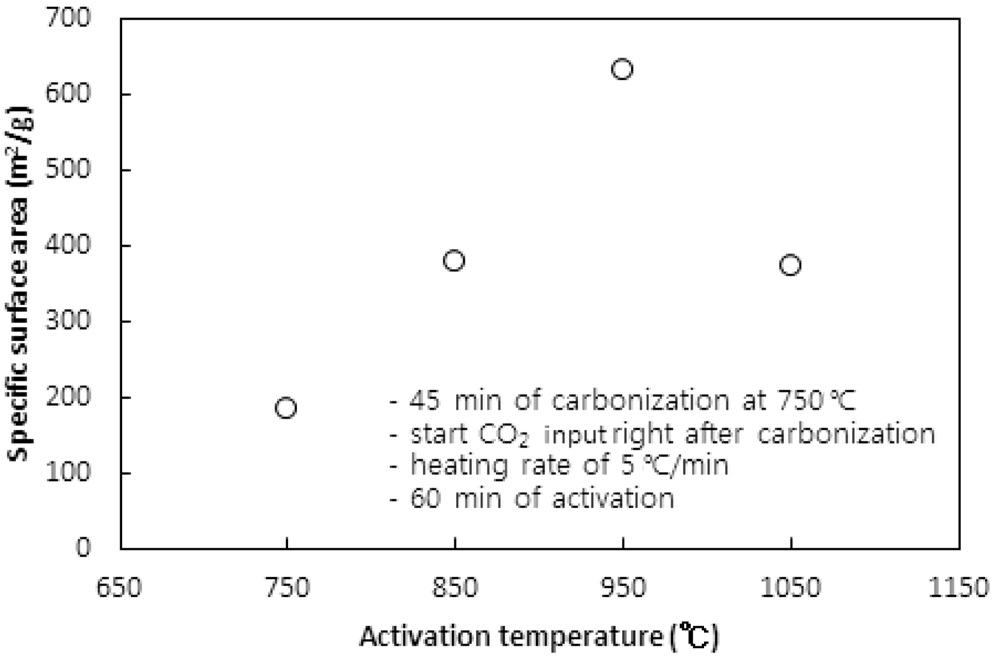
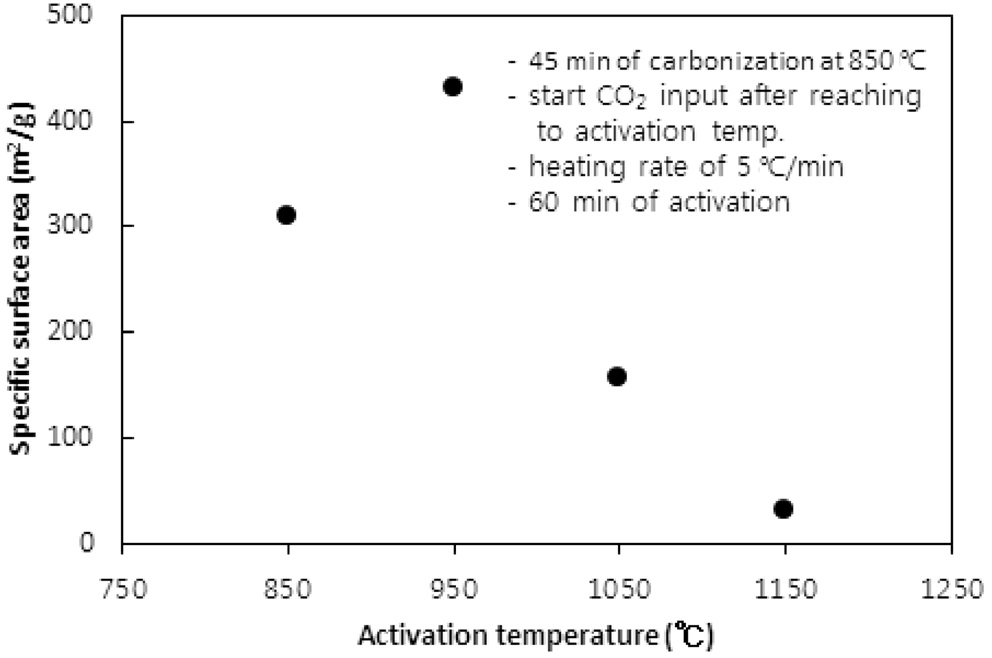
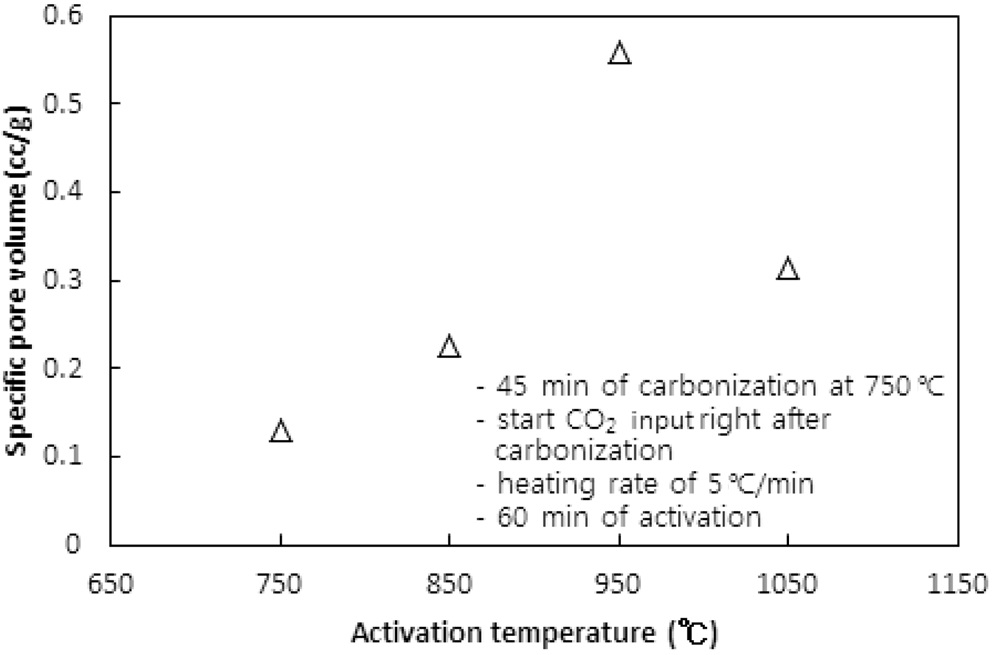
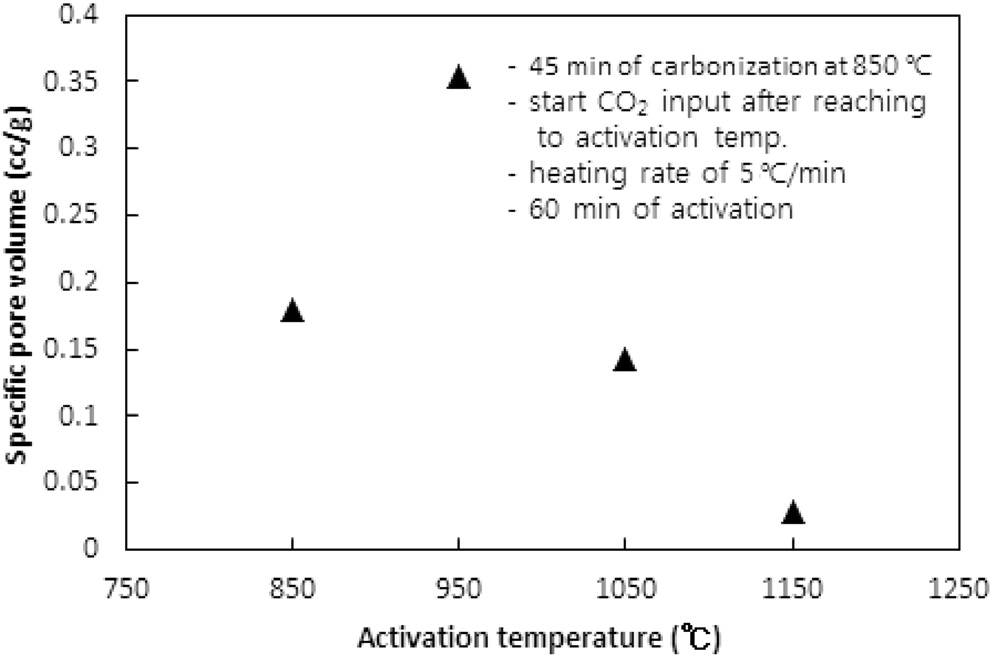
CO2의 투입방법에 따라 두 가지 방법으로 수행되었다. 즉, CO2의 투입시점을 탄화완료 직후 활성화온도로 승온 시작에서 투입하는 방법과 활성화온도로 승온 동안에는 질소를 투입하다가 활성화온도에 도달한 직후 CO2를 투입하는 방법이다. 두 방법 모두에서 탄화시간은 45분, 활성화시간은 60분, 승온속도는 5 ℃/min으로 고정했고, 탄화온도는 탄화직후 CO2를 투입 시 750 ℃ 그리고 활성화온도 도달 시 CO2를 투입 시 850 ℃로 하였다. Figure 5와 6 및 7과 8은 두 방법 각각의 비표면적과 비용적에 대한 활성화온도의 영향을 보여준다.
탄화공정 직후 활성화온도로 승온 시점부터 CO2를 투입한 경우 활성화온도가 750 ℃에서 950 ℃로 상승함에 따라 비표면적이 증가하여 950 ℃에서 본 연구에서 얻은 최대 비표면적인
631 m2/g을 얻었다. 그러나 1,050 ℃로 더 승온시켰을 경우 372 m2/g로 오히려 감소된 비표면적을 얻었다.
950 ℃까지는 산화제와 수지를 구성하는 폴리스티렌 및 DVB와의 반응속도 즉, 초미세공을 마이크로세공 및 메소세공으로 확장시키는 속도에 온도의 영향이 반영된 반면, 더 고온에서는 CO2에 의한 산화가 가속화되어 활성화공정 초기부터 형성된 마이크로세공의 일부를 메소세공과 매크로세공으로 변환시켜 비표면적과 비용적을 감소시키는 것으로 사료된다[10]. 비용적의 감소도 Figure 7에서 확인할 수 있다. 활성화온도 도달 후 CO2를 투입한 경우도 유사한 경향을 보여 950 ℃ 이상의 활성화온도에서 급격하게 감소하는 경향을 보였다.
두 경우에 동일한 활성화온도에서 탄화종료 직후부터 CO2를 투입한 경우에 더 큰 비표면적과 비용적의 변화를 보였다. 이는 승온시간을 포함하는 활성화시간이 더 길었기 때문인 것으로 사료된다. 특히 750 ℃ 45분의 탄화공정과 950 ℃ 60분의 활성화공정을 동일하게 적용했을 경우 두 방법으로 제조된 시료의 비표적과 비용적은 각각 631.2 m2/g과 0.560 cc/g 그리고 383.6m2/g과 0.3122 cc/g로 탄화종료 직후부터 CO2를 투입한 것이 더 큰 비표면적과 비용적을 보였다.
따라서 활성화온도는 950 ℃를 초과하지 않고, 탄화 직후부터 산화제가스를 투입하며 활성화온도로 승온시키는 것이 마이크로세공 및 메소세공의 생성에 적합하다.
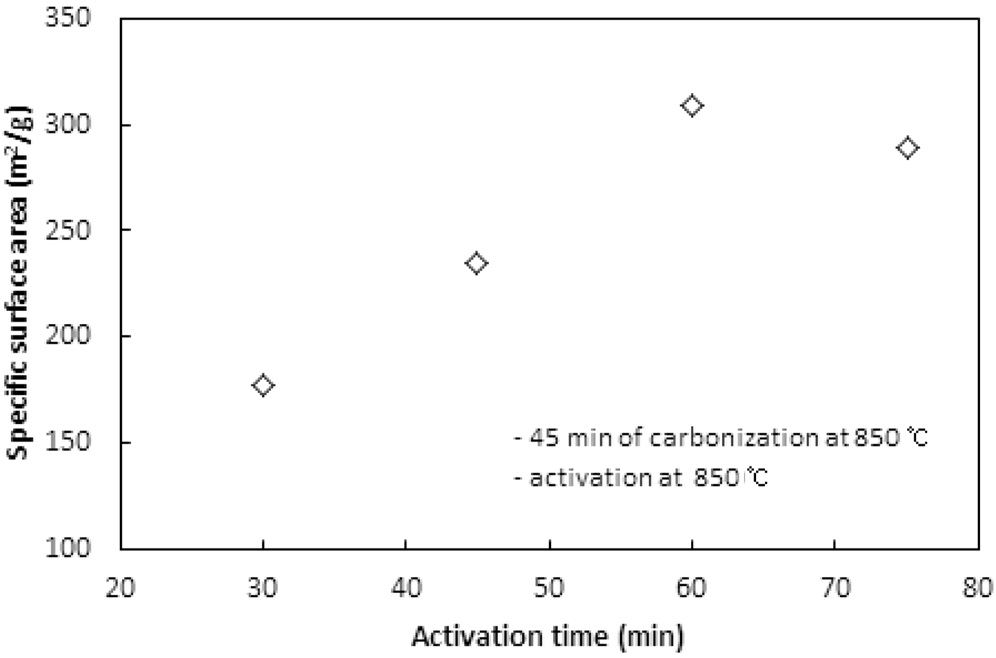
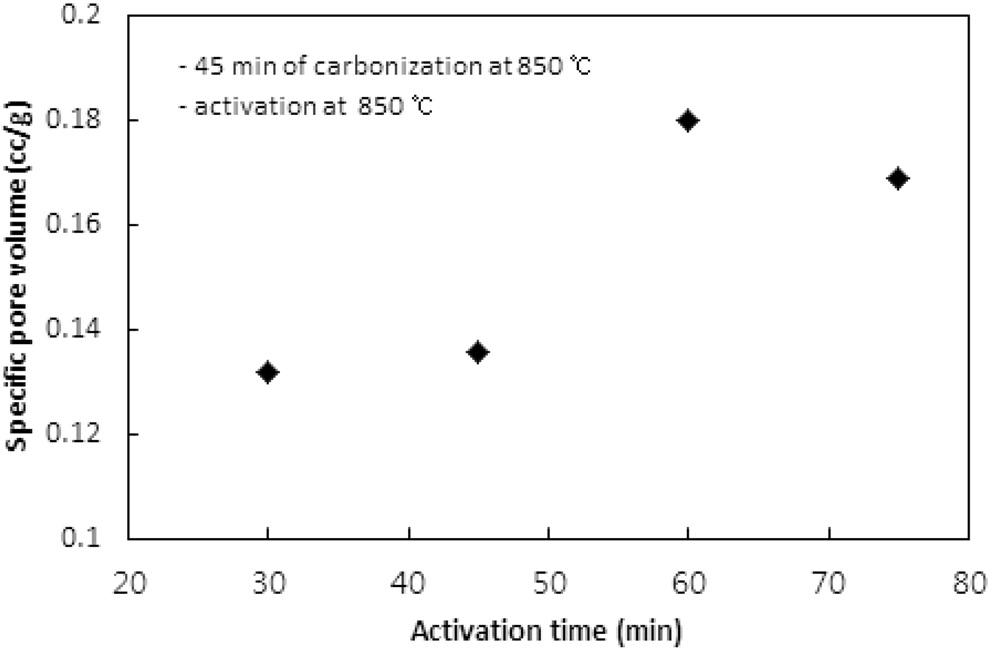
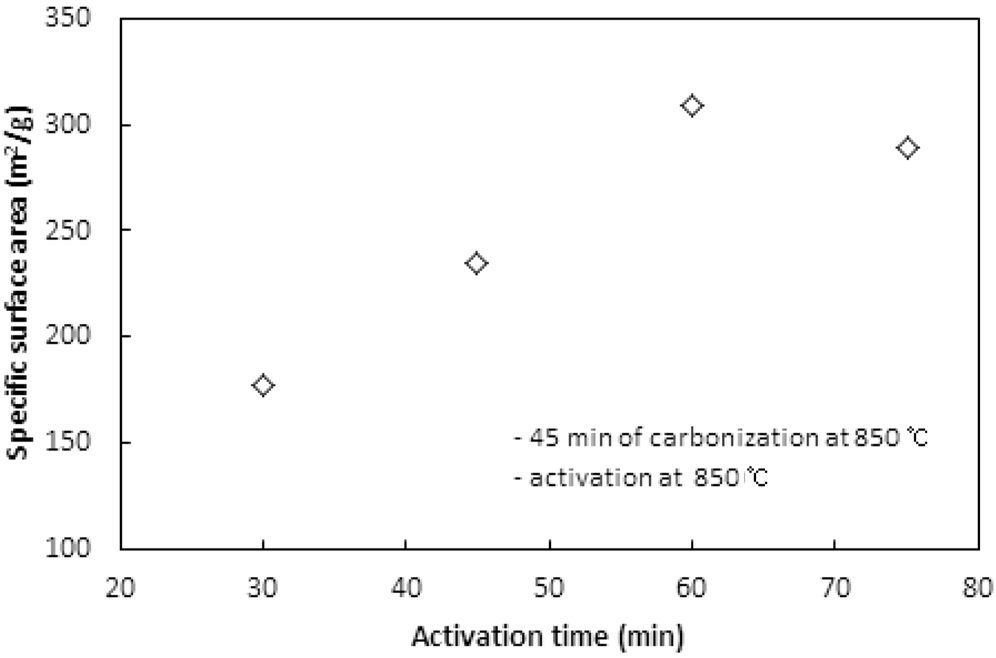
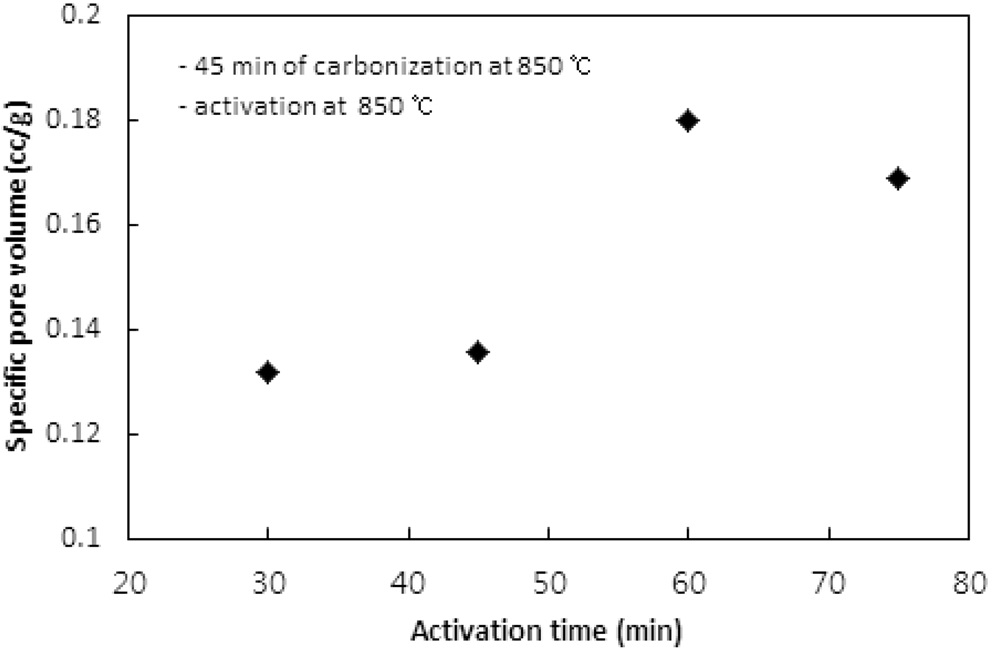
활성화온도에서 체류시간이 활성탄의 비표면적과 비용적에 미치는 영향이 실험되었다. 탄화는 850 ℃에서 45분 동안 수행되었고, 활성화는 850 ℃에서 CO2 분위기로 운전되었다. Figure 9와 10은 활성화시간에 따른 비표면적과 비용적을 보여준다. 활성화시간은 30분에서 15분씩 증가시키며 75분까지 실험하였다. 60분까지는 활성화 시간이 증가함에 따라 비표면적과 비용적이 증가하였지만, 75분에서는 60분에서의 값에 비해 약간 감소하는 경향을 보였다. 60분까지는 대부분의 산화제가 고분자와의 반응으로 탄화공정에서 생성된 초미세공을
마이크로세공과 메소세공으로 확장시키는데 사용되는 반면, 60분의 활성화시간이 초과되면서 앞서 형성된 마이크로세공과 메소세공 일부를 매크로세공으로 변화시키기 시작하여 75분에서 감소하는 경향을 보이는 것으로 판단된다.
활성화 시간은 활성화온도와 더불어 비표면적과 비용적을 제어할 수 있는 핵심 공정인자로 사용될 수 있으며, 본 연구에서 사용된 수지는 850 ℃에서 60분의 활성화시간이 적합한 것으로 나타났다.
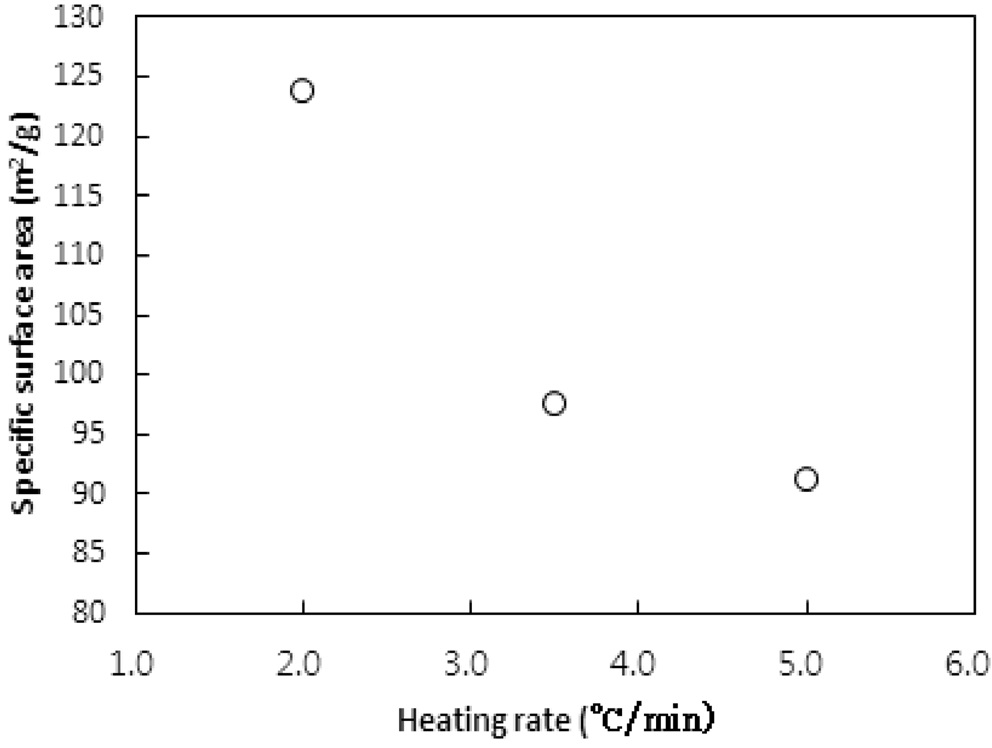
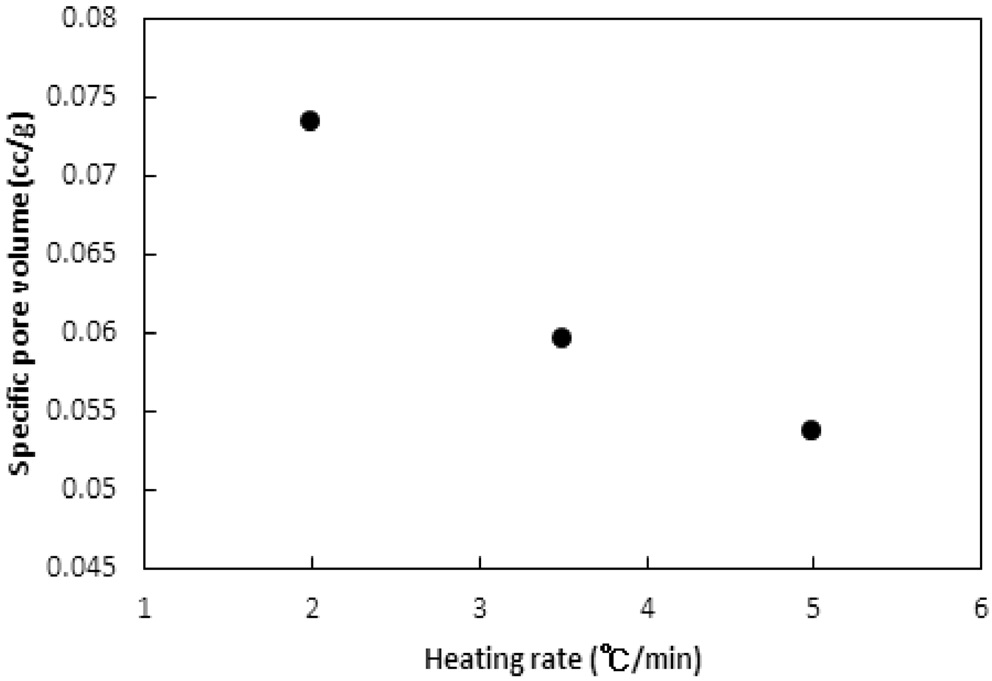
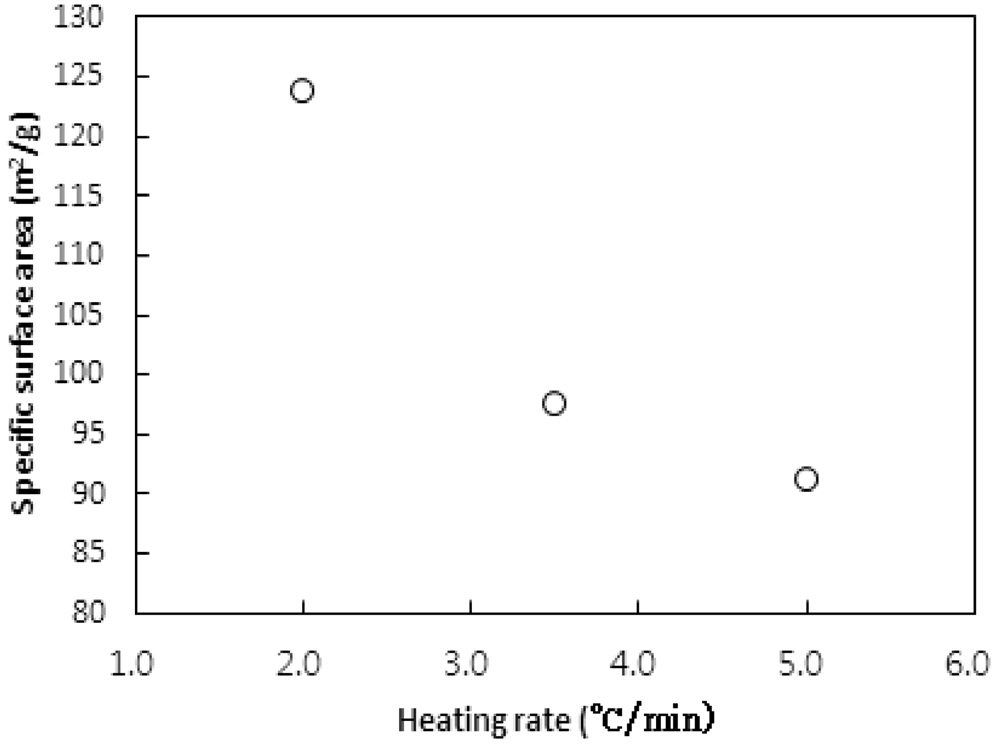
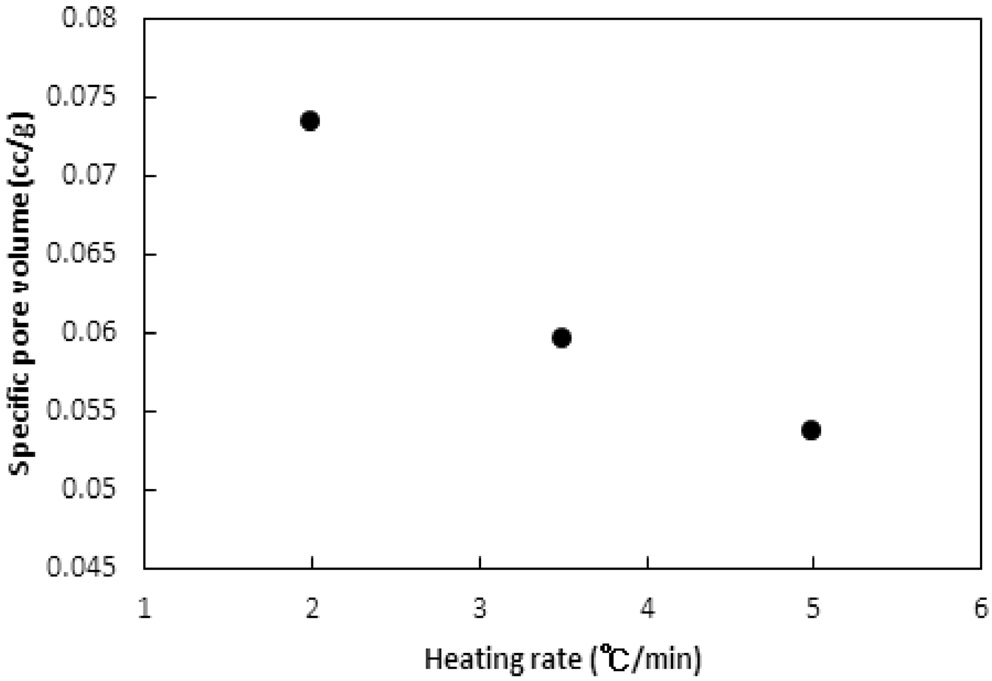
승온속도의 영향이 탄화공정에 대하여 실험되었다. 승온속도는 2, 3.5, 5 ℃/min로 변화시켰다. 탄화공정은 850 ℃에서 45분간 수행되었다. Figure 11과 12는 탄화공정에서 비표면적과 비용적에 승온속도의 영향을 보여준다.
Bratek et al.[10]은 승온속도 1, 5, 그리고 10 K/min에 대한 탄화실험에서 승온속도에 비례하여 비표면적과 비용적이 소폭 증가하였다. 하지만 본 연구에서는 비표면적과 비용적 모두 승온속도가 낮아질수록 증가하였다. 이는 탄화공정에서 주로 초미세공이 형성되며, 높은 승온속도에서 급속한 가열속도가 열수축을 촉진하여 고분자수지가 가지고 있는 초미세공 및
마이크로세공 크기의 일부 기공들이 소멸되기 때문으로 판단된다. 반대로 낮은 승온속도에서는 마이크로세공 이하의 기공들의 소멸이 감소되기 때문인 것으로 사료된다.
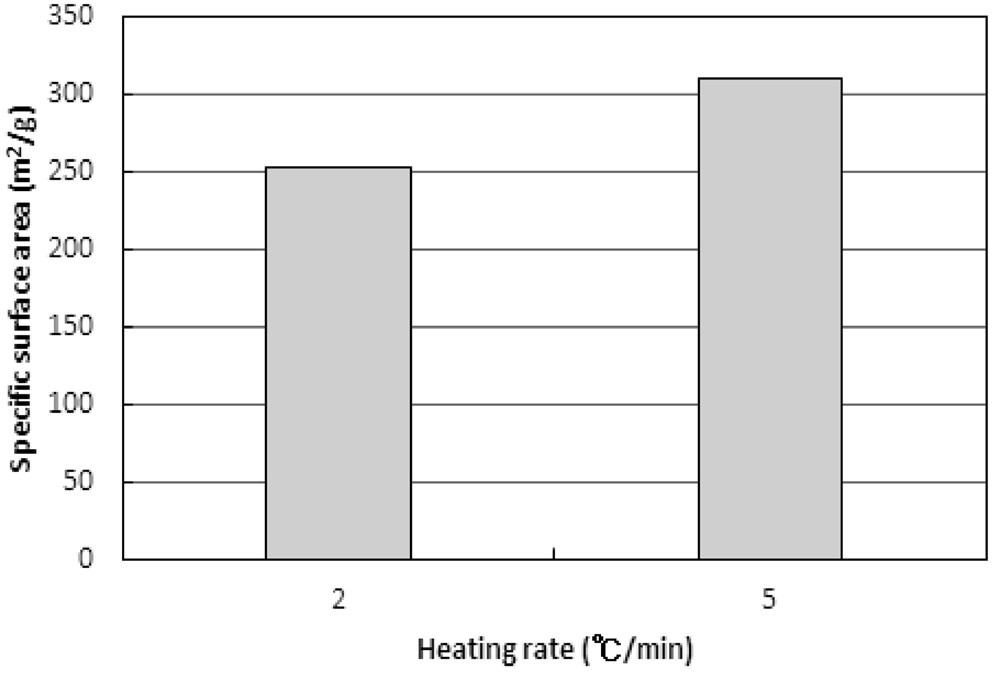
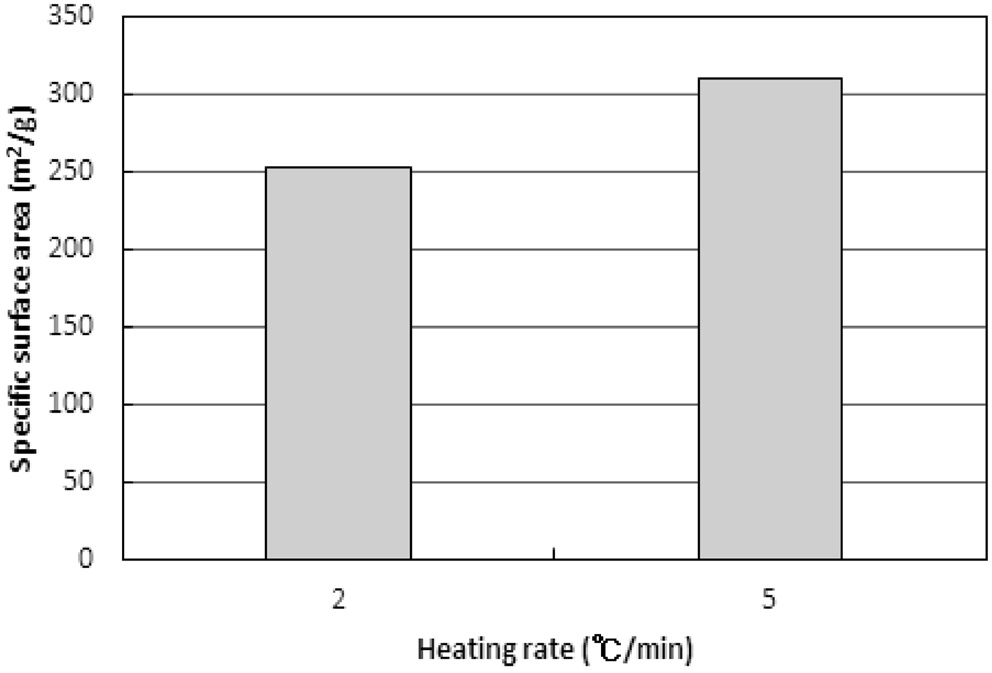
탄화물의 비표면적과 비용적이 활성화공정에서 미치는 영향을 보기 위하여 2와 5 ℃/min에서 탄화와 활성화공정을 모두 진행하였다. 탄화는 850 ℃ 45분, 활성화는 850 ℃ 60분으로 운전되었다. 그 결과는 Figure 13과 같다. 탄화공정에서 얻은 비표면적과 비용적의 결과와는 반대로 높은 승온속도로 탄화된 활성탄이 높은 비표면적과 비용적을 보였다.
이러한 결과는 탄화공정에서 형성된 탄화물의 기공을 구성하는 초미세공, 마이크로세공, 메소세공, 그리고 매크로세공의 구성비율이 승온속도에 따라 달라지기 때문으로 추측된다. 즉, 낮은 승온속도에서 탄화된 탄화물은 micropore 이상의 기공크기가 대부분이고, 높은 승온속도에서 탄화된 탄화물은 초미세공을 상당량 포함하는 것으로 추정할 수 있다. 하지만 이러한 해석은 앞서 설명된 승온속도에 따른 탄화물의 물성결과에 대한 해석에 배치된다. 두 해석에 대한 추가 연구가 필요하다.
폐이온교환수지는 장기간의 수처리공정에서 반복되는 재생을 거치며 이온교환능이 떨어져 더 이상 사용될 수 없는 상태의 수지이다. 따라서 폐이온교환수지는 수처리 중 오염이온으로 포화된 상태라 가정할 수 있다. 폐이온교환수지를 인공적으로 제조하여 탄화와 활성화공정을 수행하였다. 제조조건은 탄화 750 ℃ 45분, 활성화 950 ℃ 60분, 승온속도 5 ℃였고, 이산화탄소는 탄화공정 직후에 투입을 시작하였다.
폐이온교환수지의 인공적 제조는 대표적인 경도유발 물질인 Ca2+와 Mg2+에 대하여 각각 제조되었다. 제조방법은 Ca2+의 경우 1.0 M CaCl2용액 500 mL에 양이온교환수지 30 g을 투입한 후 간헐적으로 저어주며 12시간 이상 접촉시켜 양이온교환수지의 운전교환용량(operating capacity) 대부분을 Ca2+로 교환시켰다. Mg2+도 Ca2+와 같은 방법으로 MgCl2용액을 사용하여 Mg2+로 포화시켰다. 용액으로부터 분리한 이온교환수지는 초순수로 세척하였다.
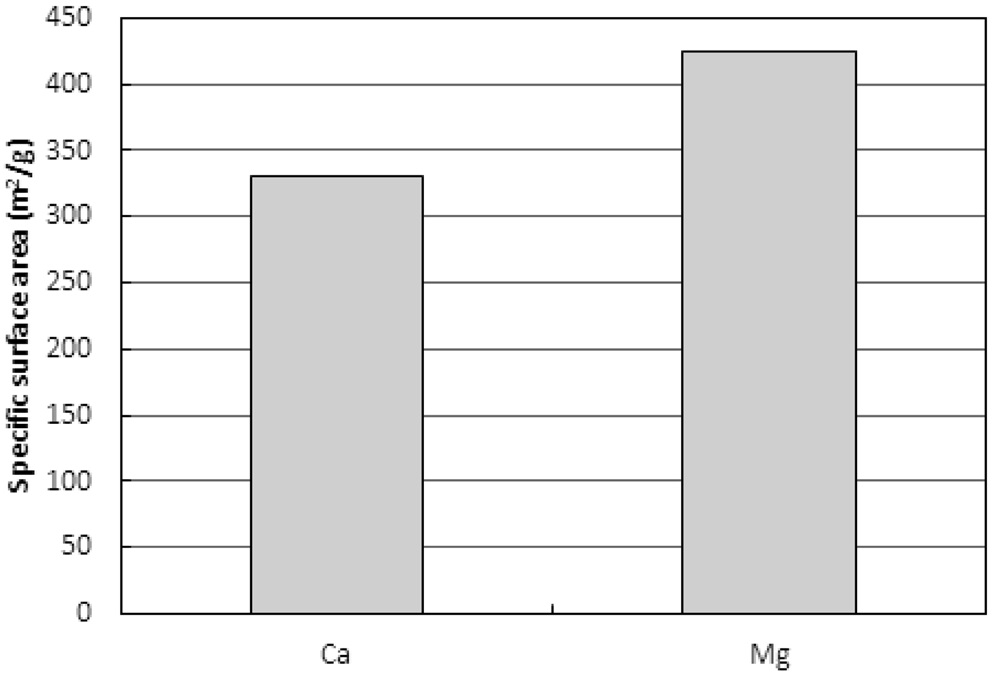
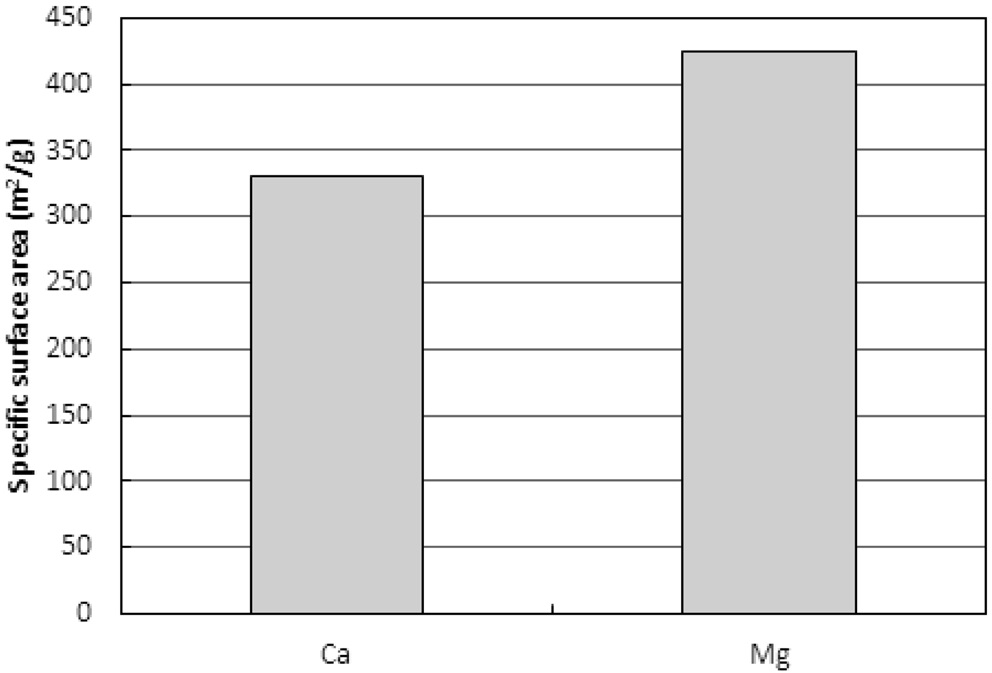
Figure 13은 두 이온들로 포화된 수지를 사용하여 활성화를 마친 활성탄의 비표면적을 보여준다. Mg2+로 포화된 수지로 제조된 활성탄이 Ca2+로 포화된 수지로 제조된 활성탄에
비해 약 30% 큰 비표면적을 나타냈다. 하지만 동일한 조건에서 새 수지로 제조된 활성탄의 비표면적은 631.2 m2/g로 Mg2+로 포화된 수지로 제조된 활성탄에 비해 약 50% 큰 값을 보였다.
Ca2+와 Mg2+ 그리고 다양한 금속이온 등과 같이 이온교환수지에 흡착된 다가이온들은 황화물(sulfide)형태 또는 금속으로 존재하며 일반 수지에 비해 더 높은 온도에서 탄화되고, 탄화공정 시 탄소층 사이에서 기둥(pillar)역할을 하여 초미세공 및 마크로세공의 열수축을 막아주는 역할을 하는 것으로 알려진다[4,13]. 이와 같이 교환된 이온이 탄화공정에서 탄화물의 비표면적과 비용적을 증가시킬 수 있겠지만 활성화공정에서 오히려 일부 기공을 막고 산화제가 기공내부로 침투하는데 저항으로 작용하며 기공표면에 붙어 비표면적과 비용적을 감소시키는 역할을 하는 것으로 예측된다. 따라서 새 수지로 제조된 활성탄에 비해 낮은 비표면적을 나타내는 것으로 사료된다. 하지만 교환된 이온의 종류와 교환량은 활성탄제조 운전인자와 더불어 제조되는 활성탄의 비표면적과 비용적을 조절할 수 있는 인자로 활용가능하다.
폴리스티렌-DVB 공중합 고분자인 이온교환수지를 탄소원으로 활성탄제조 시 탄화와 활성화공정에서 운전인자가 활성탄의 물성인 비표면적과 비용적에 미치는 영향이 실험적으로 연구된 결론은 다음과 같이 요약된다.
1) 탄화공정에서 탄화온도가 증가함에 따라 열수축의 증가로 탄화물의 비표면적과 비용적이 선형적으로 감소하였지만, 탄화물의 수율은 변화가 없었다. 또한 탄화시간이 길어짐에 따라 열수축으로 인하여 비표면적과 비용적이 감소하는 경향을 보였고, 산화제의 지속적인 분해반응으로 탄화수율이 소폭 감소하였다.
2) 활성화공정에서 950 ℃까지는 활성화온도가 증가함에 따라 산화제 반응속도에 대한 온도의 영향으로 마이크로세공의 생성이 촉진되어 비표면적과 비용적은 증가하였지만, 그 이상의 온도에서는 지나친 산화반응으로 생성된 마이크로세공이 메소세공 및 매크로세공으로 변환되어 감소하였다. 산화제의 투입은 탄화직후부터 투입하는 것이 비표면적 및 비용적 증가에 유리했다. 한편 활성화시간의 경우 60분까지는 비표면적과 비용적이 증가하였지만, 그 이상의 활성화는 지나친 산화로 감소하였다.
3) 탄화공정에서 승온속도가 낮을수록 탄화물의 비표면적과 비용적은 증가하였다. 승온속도가 높을수록 탄화 시 열수축이 심해져 비표면적과 비용적이 감소하는 것으로 해석된다. 그러나 위 탄화물을 활성화한 경우 반대로 높은 승온속도에서 제조된 활성탄이 더 높은 비표면적과 비용적을 보였다. 정확한 해석을 위한 추가연구가 필요한 것으로 사료된다.
4) Ca과 Mg이온으로 포화된 수지로 활성탄을 제조한 결과 새 수지에 비해 기둥효과로 높은 비표면적을 보일 것으로 예상했지만 오히려 낮은 비표면적을 보였다. 운전인자와 함께 교환된 이온의 종류와 양은 활성탄 물성조절을 가능하게 해주는 역할을 할 것으로 판단된다.

![Physicochemical properties of CMP 16[12]](http://oak.go.kr/repository/journal/12627/CJGSB2_2013_v19n3_279_t001.jpg)