About five years ago, a national project named the Korean Tree of Life (KTOL) Project was initiated in Korea, mimicking the Tree of Life (ToL). Throughout this project, many taxonomic researchers had the chance to revise, rename, and uncover by comparing the sequence-based identity to the traditional morphological data and to sequence databases such as GenBank. Furthermore, the sequence information was also utilized to complement the phylogenetic relationships of given taxonomic groups occurring in South Korea to those occurring outside the Korean peninsula, although the information is limited to the perspective of world taxonomic diversity and a large sequencebased analysis, particularly considering current trends (e.g., Mutanen et al., 2010; Regier et al., 2009; Wahlberg et al., 2009).
A recent ecological overview has shown ecological diversity in Korean butterflies, including Nymphalidae, in terms of biogeographic origin, distributional range, habitat characteristics, voltinism, and so on (Kim, 2012). According to Kim (2012), ~80 among ~90 species (38 genera in seven subfamilies) occurring in South Korea originated from a northern region, whereas only ~10 species, including the typical southeastern genus Parantica, originated from a tropical region. With regard to voltinism, ~60 species are univoltine, whereas the remaining species are either bivoltine, trivoltine, or multivoltine (Kim, 2012). Due mainly to such different voltinisms, differences in seasonal wing morphology, body size, and the overwintering stage are known to be prevalent.
With regard to taxonomic perspective, the Nymphalidae along with other butterflies occurring in South Korea were further welllisted by several important earlier studies since the beginning work by foreign scientists (see Kim, 2012). The subsequent majority of Nymphalidae research in South Korea has focused on introduction of individual species in illustrated books (e.g., Kim, 2002), finding and listing new species through morphological analysis (e.g., Joo et al., 1997; Lee and Takakura, 1981; Park, 1987), and ecological investigation of a limited number of species (e.g., Kim, 2012).
For phylogenetic perspective, a report on the familial relationships of Papilionoidea was nearly a unique one that included a substantial number of species that occur in South Korea (Kim et al., 2010). Even so, much more research effort is required to examine taxonomic diversity in South Korea. On the other hand, in the case of world perspective on phylogeny, several studies analyzed various subgroups of Nymphalidae along with other butterflies using diverse molecular markers and morphological characters (Ehrlich, 1958; Ehrlich and Ehrlich, 1967; DeVries et al., 1985; Martin and Pashley, 1992; Freitas and Brown Jr, 2004). A report by Wahlberg et al. (2009) is noteworthy in that it compiled a data matrix of 10 genes and 235 morphological characters with taxonomic representation of all subfamilies, tribes, and subtribes of Nymphalidae. With regard to subfamilial relationships, they found Libythinae to be the most basal lineage and placed Danainae at the next branch, presenting the other subfamilial relationships as (Satyrinae + ((Nymphalinae + Apaturinae) + (Limenitidinae + Heliconiinae))).
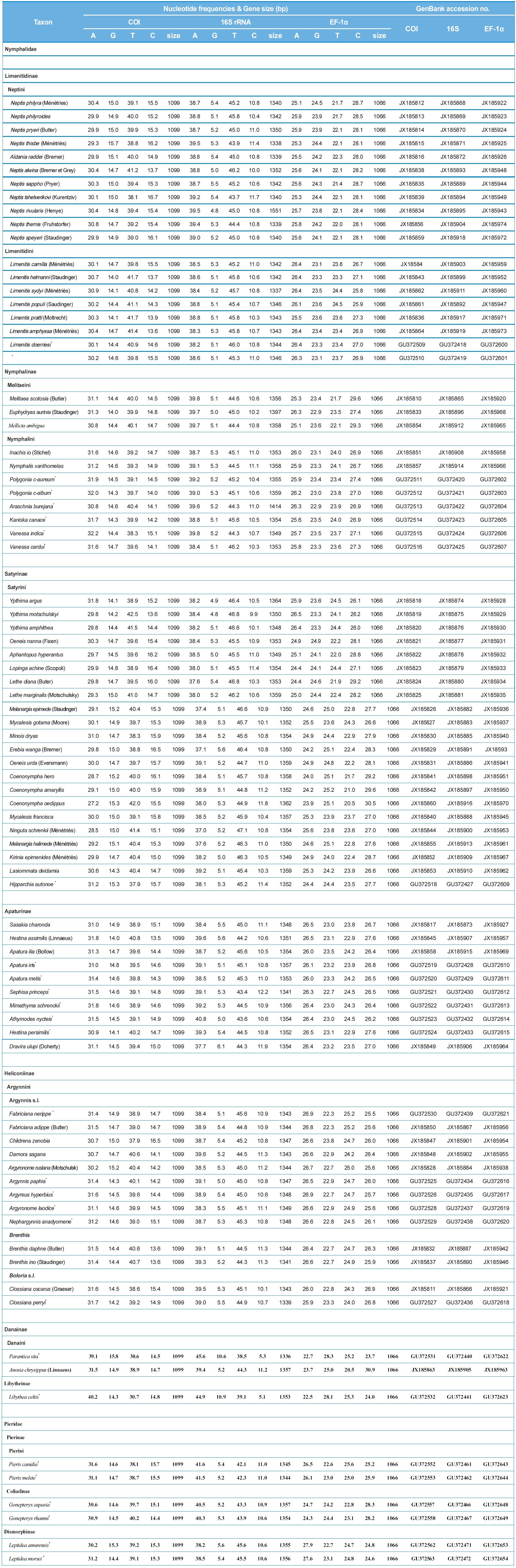
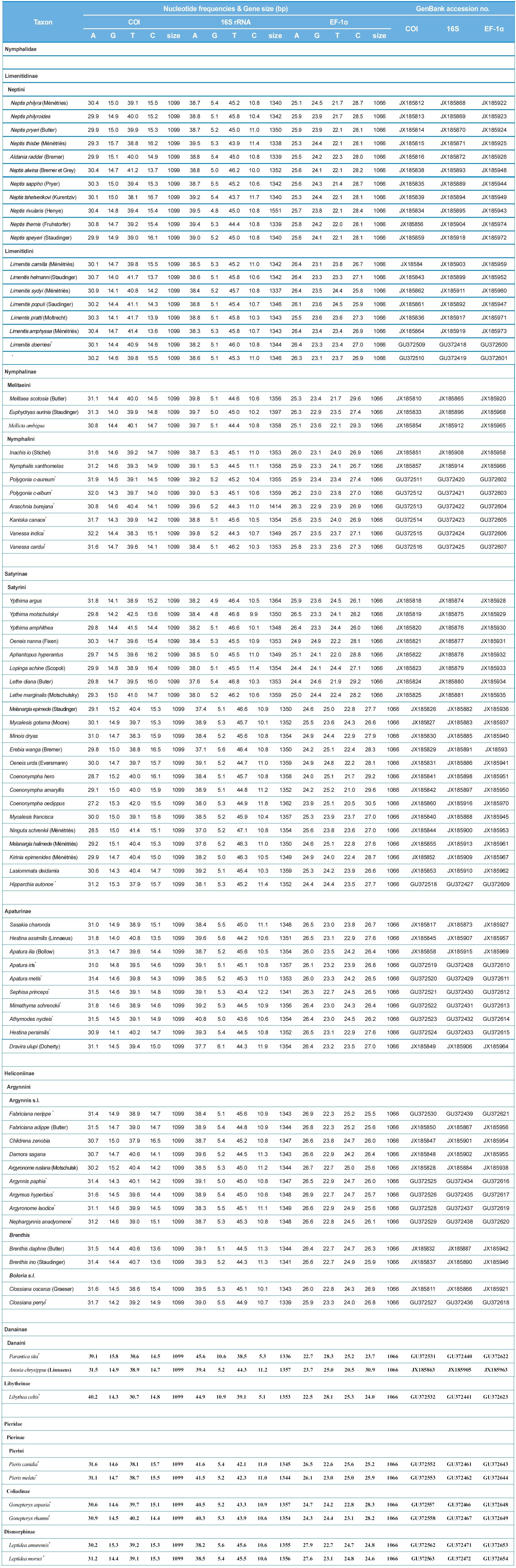
As part of the KTOL project, in this study, 78 species of Nympahlidae were analyzed for their cytochrome oxidase subunit I (COI), 16S ribosomal RNA (16S rRNA), and elongation factor-1 α (EF-1α), each of which spans 3,501?3,716 bp. Twenty-three species that were previously reported by Kim et al. (2010) and 55 species that were newly sequenced in this study together belong to 47 genera in seven subfamilies, accounting for 86% of Nympalidae occurring in South Korea (Kim, 2012). This sequence information including DNA barcode regions is expected to be helpful for species identification and for the inference of international relationships of the Nymphalidae occurring in South Korea, providing the basal data for further research. Although limited, this study contains the most diverse nymphalid species occurring in South Korea.
Seventy-eight adult butterflies belonging to family Nymphalidae and representing 47 genera in seven subfamilies were used in this study (Table 1). Among them, 23 species were taken from the previous study (Kim et al., 2010). For phylogenetic analysis, six species of Pieridae representing three subfamilies were taken from Kim et al. (2010) for outgroups in consideration of previous studies (Ackery, 1988; Robbins, 1988; de Jong et al., 1996; Campbell et al., 2000). Voucher specimens were deposited in the National Institute of Biological Resources, Incheon, Republic of Korea.
After collection from the field, samples were frozen at -70℃ until used for molecular analyses. Total DNA was extracted with a Wizard™ Genomic DNA Purification Kit according to the manufacturer's instructions (Promega, USA).
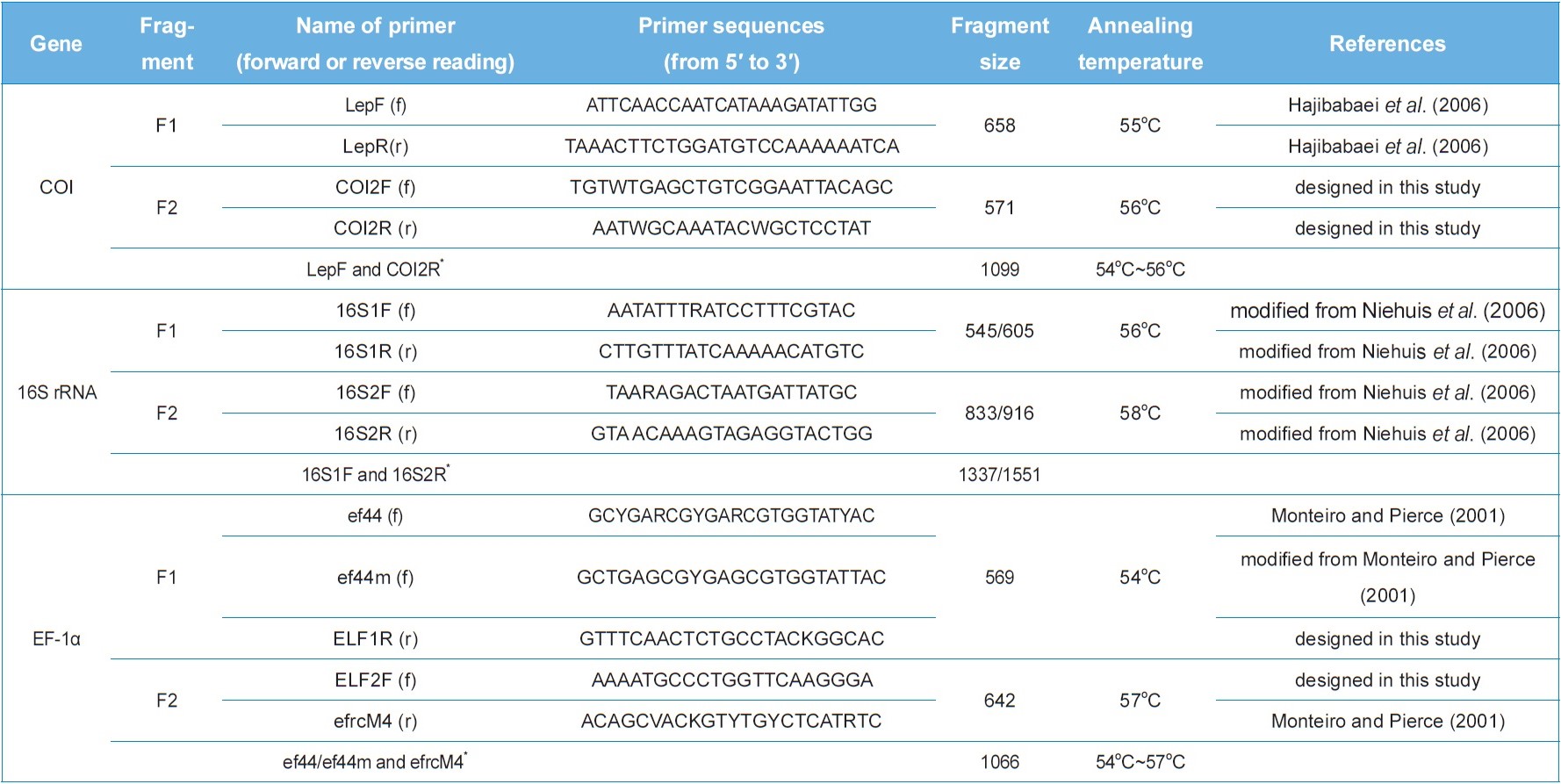
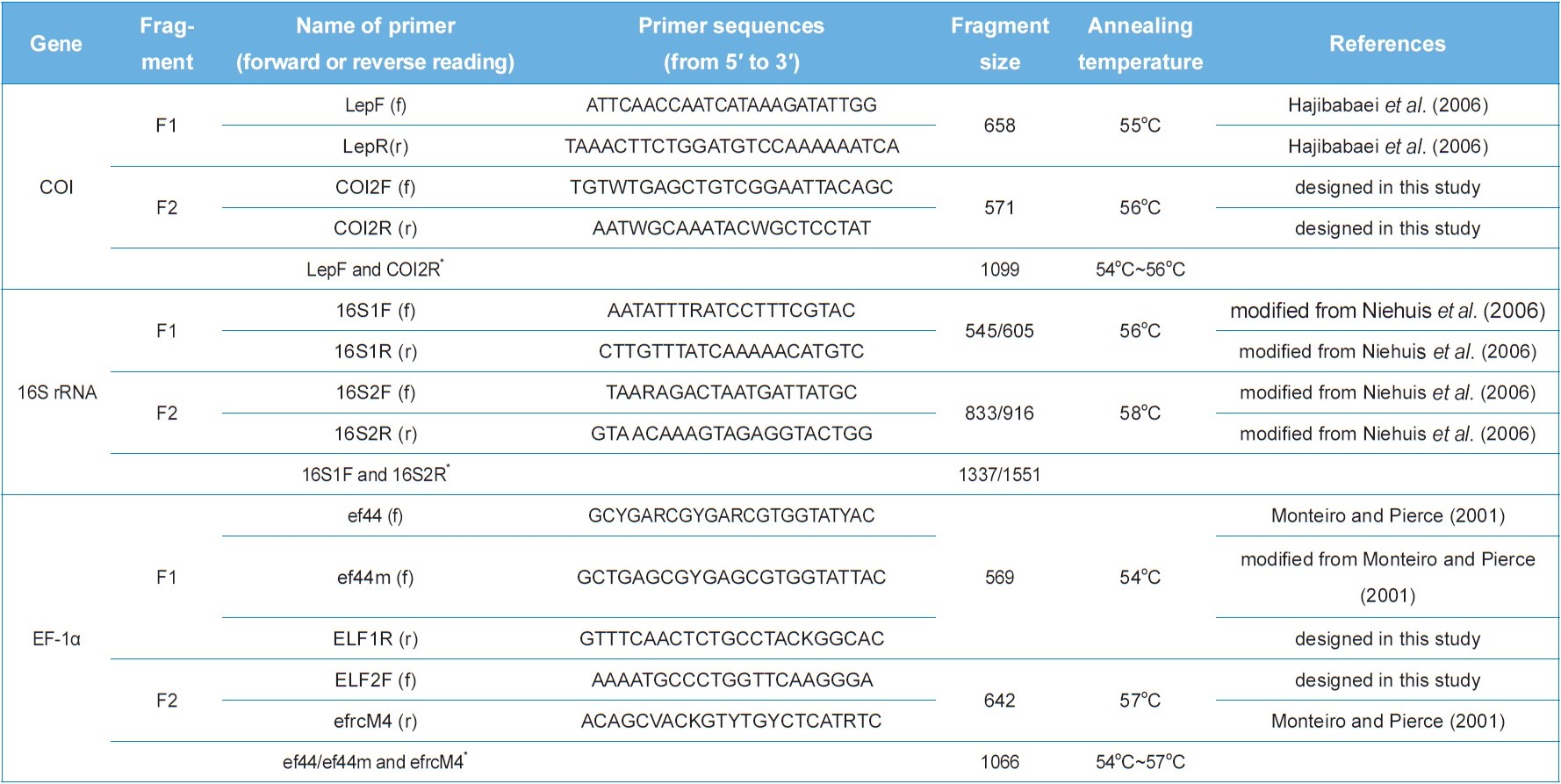
To sequence the mitochondrial COI, 16S rRNA, and nuclear EF-1α genes, each gene was amplified into two independent fragments in most cases for easy processing. All PCR products were sequenced either directly or after cloning. The details of primer sets used for the amplification of each fragment are provided in Table 2. They were either from previous studies or newly designed. PCR amplification was conducted using AccuPower® PCR PreMix (Bioneer, Korea) under the following conditions: initial denaturation for 7 min at 94℃, followed by 35 cycles of 60 s at 94℃, 60 s at 54?58℃, and 2 min at 72℃, with a subsequent final 7-minute extension at 72℃. To confirm successful DNA amplification, electrophoresis was conducted using 0.5× TAE buffer on 0.5% agarose gel. The PCR product was subsequently purified with a PCR purification Kit (QIAGEN, Germany). Cloning was carried out using pGEM-T Easy vector (Promega, USA). The resultant plasmid DNA was isolated using a Wizard Plus SV Minipreps DNA Purification System (Promega, USA). DNA sequencing was conducted using the ABI PRISM® BigDye® Terminator v3.1 Cycle Sequencing Kit with an ABI 377 Genetic Analyzer (PE Applied Biosystems, USA). All the products were sequenced from both strands.
Nucleotide sequences for each gene were aligned using MAFFT ver. 6 (Katoh et al., 2002), with the gap opening penalty set to 1.53 and the offset value (equivalent to gap extension penalty) set to 0.5. Subsequently, the well-aligned blocks were selected using GBlocks 0.91b (Castresana, 2000), with the maximum number of contiguous non-conserved positions set to four for the COI and EF-1α genes. The 16S rRNA gene was subjected to GBlocks analysis using parameters optimized for rDNA alignments (minimum length of a block as five, allowing gaps in the half position) (Massana et al., 2004). Considering the general trend towards utilization of combined information including previous phylogenetic studies on Papilionoidea (Wahlberg et al., 2005, 2009; Savard et al., 2006), phylogenetic analysis was performed using the combined three genes. Bayesian inference (BI) and Maximum likelihood (ML) methods were employed for phylogenetic reconstruction. For the BI analysis, the dataset was either unpartitioned or divided into three partitions based on each gene to assess the effect of data partitioning. The average sequence divergence (%) within a tribe, subfamily, and family for individual and combined gene sequences were estimated under an uncorrected p-distance model using MEGA5 (Tamura et al., 2011).
Substitution model selection was performed by comparison of Akaike Information Criterion (AIC) scores (Akaike, 1974), calculated using the Modeltest ver. 3.7 (Posada and Crandall, 1998). In each gene and combined gene sequences, the GTR (Lanave et al., 1984) + I + G model was selected and applied for the BI and ML methods. The ML analyses were conducted using PHYML (Guindon et al., 2005), specifying the number of substitution rate categories as four, and the proportion of invariable site and the number of substitution sites set as the values obtained from the model test with the starting tree set as a BIONJ distance-based tree. The confidence values were evaluated via the bootstrap test with 1,000 iterations. The BI analyses were conducted using MrBayes ver. 3.1 (Huelsenbeck and Ronquist, 2001). When the partition option was employed, the model chosen by Modeltest (Posada and Crandall, 1998) was applied to each partition unlinked. Two independent runs of four incrementally heated Markov Chain Monte Carlo (MCMC) chains (one cold chain and three hot chains) were simultaneously run for one-to-three million generations depending on the dataset, with sampling conducted every 100 generations. The convergence of MCMC, which was monitored by the average standard deviation of split frequencies, reached below 0.01 within one-to-three million generations depending on the dataset, and the initial 25% of the sampled trees were discarded as burnin. The confidence values of the BI tree are presented as the Bayesian posterior probabilities (BPP).
The sequence lengths of 78 nymphalid and six pierid species were 1,099 bp for COI, 1,336?1,551 bp for 16S rRNA, and 1,066 bp for EF-1α (Table 1). Both COI and EF-1α did not have any insertions/deletions. The concatenated sequences of the three genes ranged 3,501?3,716 bp among species. However, the conserved blocks selected by GBlocks analysis (Castresana, 2000) eventually gave a total of 3,404 bp, composed of 1,243 bp from 16S rRNA (64% of original sequences including 92 indels),
1,097 bp from COI (99% of original sequences), and 1,064 bp from EF-1α (99% of original sequences).
The average A/T content of COI, 16S rRNA, and EF-1α were 70.6%, 84.2%, and 49.2%, respectively (Table 1). This result provided evidence of the A/T bias in mitochondrial genes, but not for nuclear genes. Between the two mitochondrial genes, the 16S rRNA gene showed much higher A/T content than that of COI gene. The A/T content of protein coding genes and 16S rRNA in Lepidoptera was approximately 75.7%?81.5% and 81.4%?85.1%, respectively (Kim et al., 2011), indicating somewhat higher A/T content in 16S rRNA.
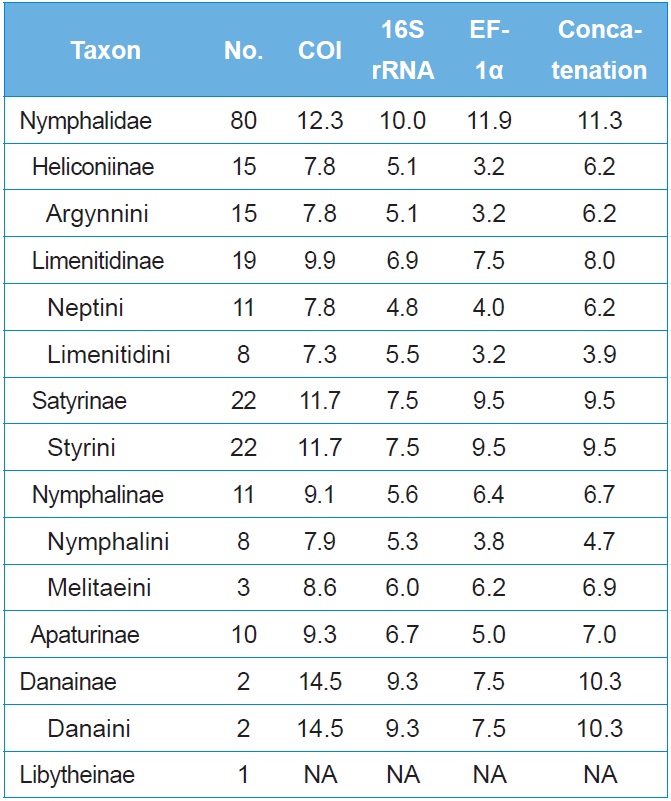
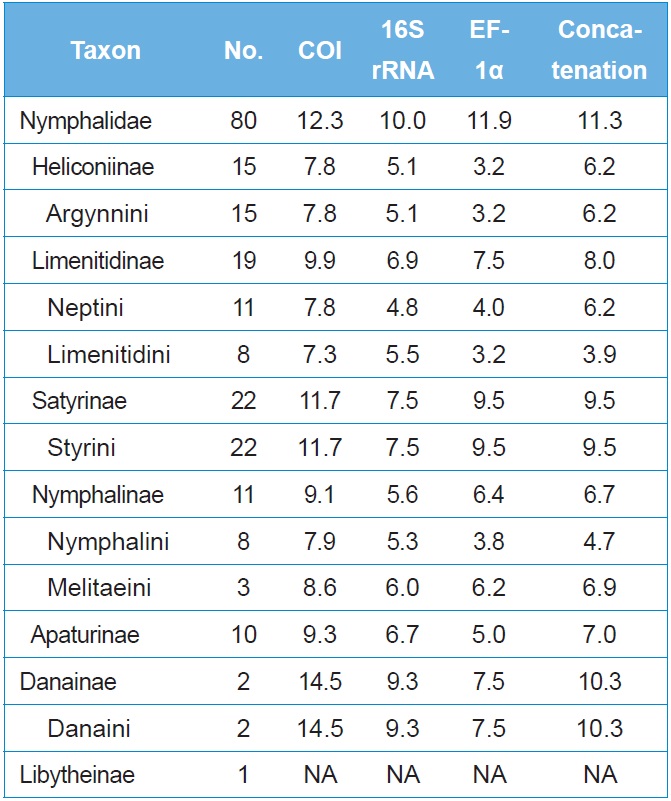
The average sequence divergence of within-subfamily ranged from 7.8%?14.5% in COI, 5.1%?9.3% in 16S rRNA, and 3.2%?9.5% in EF-1α, with the concatenated divergence ranging from 4.7%?10.3% (Table 3). In six nymphalid subfamilies, the concatenated divergence ranked in the following order: Danainae (10.3%), Satyrinae (9.5%), Limenitidinae (8.0%), Apaturinae (7.0%), Nymphalinae (6.7%), and Heliconiinae (6.2%). The same rank of sequence divergence was also found in the individual COI and 16S rRNA genes, but that for the EF-1α gene ranked in the order of Satyrinae, Danainae, Limenitidinae, Nymphalinae, Apaturinae, and Heliconiinae (Table 3), indicating different modes of evolution between the two types of genes.
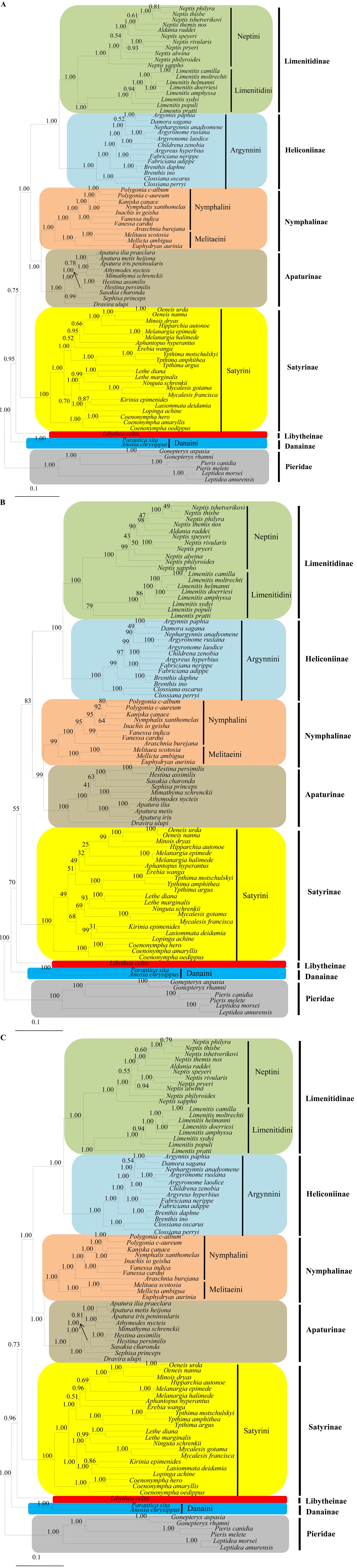
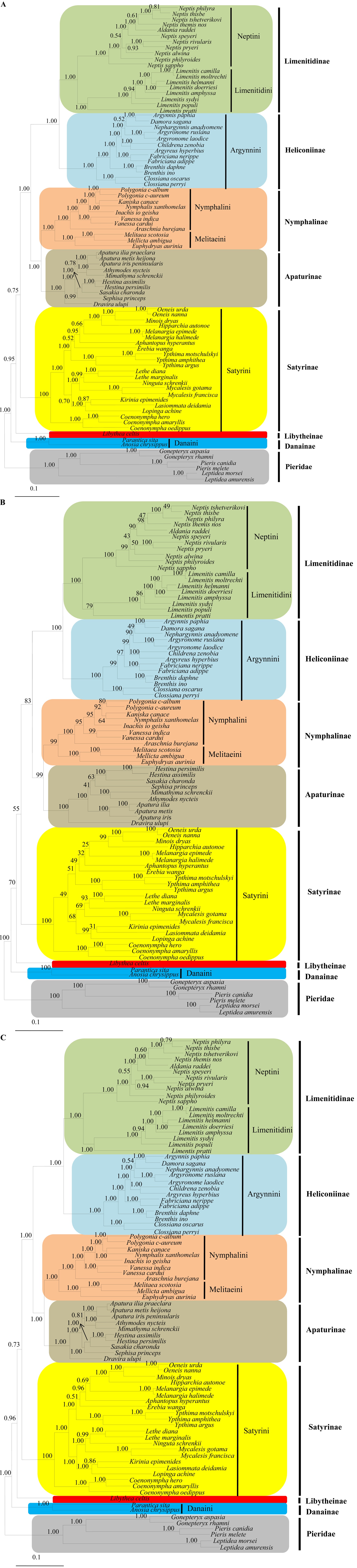
All the analyses based on concatenation of the COI gene plus the EF-1α and 16S rRNA genes, regardless of analytical methods and partitioning strategies, concordantly recovered all nymphalid subfamilies and tribes as monophyletic groups with high nodal supports (Fig. 1). This result was consistent with the latest phylogenetic study using combined molecular and morphological data (Wahlberg et al., 2009) and early morphological studies (Ehrlich, 1958; Ehrlich and Ehrlich, 1967).
With regard to subfamilial relationships, all analyses, regardless of analytical methods, supported Danainae as the most basal lineage of Nymphalidae, placing it as the sister to Libytheinae and the remaining subfamilies with high nodal supports (1.00 by BI and 100% by ML analyses) (Fig. 1). This result has never been proposed in previous studies, except for that by Kim et al. (2010), wherein the concatenated COI, 16S rRNA, and EF-1α genes were also used for phylogenetic reconstruction among true butterfly families. Several phylogenetic studies based on morphological (Ehrlich and Ehrlich, 1967; Freitas and Brown Jr, 2004) and/or molecular data (Wahlberg et al., 2003, 2009) placed Danainae as the sister to the remaining subfamilies of Nymphalidae, but placed Libytheinae as the most basal lineage. Such placement has been widely supported by morphological characteristics, such as lack of reduced female foreleg, an apomorphic feature found in all other Nymphalidae (Ehrlich and Ehrlich, 1967; Scott, 1984; Freitas and Brown Jr, 2004) and/or molecular data (Martin and Pashley, 1992; Wahlberg et al., 2009). Furthermore, host plant and geographic distribution also suggested that this subfamily was an outgroup of the remaining Nymphalidae, as the most basal subfamily (Ackery, 1984; Freitas, 1999; Vane-Wright, 2003). Thus, the placement of Danainae at the most basal branch, instead of Libytheinae in our study, is likely a consequence of limited sampling.
The subfamily Satyrinae was consistently placed as the next basal lineage after Libytheinae and Danainae in all analyses with moderate and low supports by BI (0.73 and 0.75) and by ML analyses (55%), respectively (Fig. 1), providing the subfamilial
relationships (((Limenitidinae + Heliconiinae) + (Nymphalinae + Apaturinae)) + Satyrinae). This relationship is concordant with that proposed by Wahlberg et al. (2009), but not to those obtained by several earlier morphology-based studies (Ehrlich, 1958; DeVries et al., 1985; Freitas and Brown, 2004).
The sister relationships between Nymphalinae and Apaturinae were strongly supported in the present study (1.00 by BI and 100% by ML analyses). This result was consistent with that revealed by Ehrlich and Ehrlich (1967) and Wahlberg et al. (2009), but differed from that by Martin and Pashley (1992), wherein 28S and 18S rRNA data were used and were consistent with the results demonstrated by Freitas and Brown Jr (2004), wherein morphological characters from all stages, such as the color of eggs, caudae of larvae, and wing patterns of adults were used. Martin and Pashley (1992) demonstrated neither Apaturinae nor Nymphalinae to be monophyletic. In the case of Apaturinae, one genus (Doxocopa) was placed as the sister to Danainae, leaving another genus (Asterocampa) as the sister to this group. In the case of Nymphalinae, one genus (Vanessa) was placed as the sister to the group composed of ((Danainae + Doxocopa) + Asterocampa). Freitas and Brown (2004) proposed Apturinae as a monophyletic group, but placed it as the sister to Satyrinae, leaving them as the most recently derived lineages of Nymphalidae. However, Nymphalinae was also recovered as a nonmonophyletic group, placing the majority of Nymphalinae (Nymphalini, Kallimini, and Melitaeini) as sisters to Heliconiinae, while the single tribe Coeini was placed as the sister to Limenitidinae. The sister relationship between Limenitidinae and Heliconiinae was supported with the highest nodal supports (1.00 by BI and 100% by ML analyses), consistent with that revealed by Wahlberg et al. (2009).
Overall, the phylogenetic analyses in the present study yielded each subfamily and tribe as a monophyletic group, and the subfamilial relationships within Nymphalidae as (((((Limenitidinae + Heliconiinae) + (Nymphalinae + Apaturinae)) + Satyrinae) + Libytheinae) + Danainae).
In the tribe Satyrini of Satyrinae, six of 14 genera (Oeneis, Melanargia, Ypthima, Lethe, Mycalesis, and Coenonympha) represented by more than one species were all consistently shown to be monophyletic groups in all analyses (Fig. 1). Concordantly in all analyses, Coenonympha (three species) was placed as the most basal lineage of Satyrini with high nodal supports (1.00 by BI and 100% by ML analyses), consistent with that reported by Martin et al. (2000), wherein 16S rRNA was used. The remaining genera were subdivided into two groups, forming Ypthima as the sister to the group composed of (((((Oeneis + Minois) + Eumenis) + Melanargia) + Aphantopus) + Erebia) on one hand (1.00 by BI and 51% by ML analyses), and the group composed of ((Kirinia + Lasiommata) + Pararge) as the sister to (Mycalesis + (Lethe + Ninguta)) on the other hand (1.00 by BI and 68% by ML analyses) (Fig. 1). This result conflicted with previous studies. For example, Martin et al. (2000) reported the genus Lasiommata at the most basal branch and the genus Pararge at next branch, placing the genus Coenonympha as the sister to the remaining Satyrini based on concatenated mitochondrial 16S rRNA and ND1 genes. Pena et al. (2011) using substantially large molecular data reported that the group (((Mycalesis + Lethe) + Pararge) + Coenonympha) was basal to the Satyrini. Probably, an expended sampling and sequences for the Satyrini might be needed to better understand the relationships within the Satyrini that occur in South Korea.
The genera Apatura, Mimathyma (= Athymodes), and Hestina, that were sampled for more than one species in Apaturinae were all consistently revealed as monophyletic groups with high nodal supports (1.00 by BI and 100% by ML analyses) (Fig. 1). All analyses consistently revealed Dravira as the most basal lineage of Apaturinae, forming the remaining genera as a monophyletic group with high nodal supports (1.00 by BI and 100% by ML analyses), consistent with previous studies (Ohshima et al., 2010). However, the internal relationship within the remaining genera fluctuated depending on the analytical method. BI analyses, regardless of data partitioning, showed the group composed of (Apatura + Mimathyma) as the sister to the group composed of (Sephisa + (Hestina + Sasakia)), whereas the ML analysis supported Apatura as the sister to (Mimathyma + (Sephisa + (Hestina + Sasakia))), although the nodal supports for the latter grouping were obviously lower (Fig. 1). Nevertheless, both relationships conflicted with previous studies, which either suggested Apatura as the sister to Hestina + Sasakia on the basis of a single mitochondrial gene (Zhang et al., 2008) or Sephisa as the sister to Apatura + Mimathyma on the basis of combined mitochondrial and nuclear genes (Ohshima et al., 2010). The Apaturinae is one of the unresolved nymphalid groups, consisting of 430 described species belonging to 20 genera. However, the taxon-specific morphologies, such as an extremely elongated phallus and saccus in male genitalia that are absent from other butterfly species, support the monophyly of Apaturinae (Le Moult, 1950; Shirozu and Saigusa, 1971; Ackery et al., 1999). As has been supported by molecular data (Wahlberg et al., 2003; Zhang et al., 2008; Ohshima et al., 2010) and combined molecular and morphological data (Wahlberg et al., 2005, 2009), the monophyly of this subfamily is also supported in our study (Fig. 1).
In the subfamily Nymphalinae, the tribe Nymphalini (seven genera) and the tribe Melitaeini (three genera) each were well recovered as monophyletic groups with strong support in all analyses (Fig. 1). Two genera, Polygonia and Vanessa, represented by more than one species in the Nymphalini, were consistently recovered as monophyletic groups in all analyses. Furthermore, all analyses concordantly supported the phylogenetic relationships ((((((Nymphalis + Kaniska) + Polygonia)) + Inachis) + Vanessa) + Araschnia) within Nymphalini and ((Melitaea + Mellicta) + Euphydryas) within Melitaeini (Fig. 1). Placed as the sister to Vanessa, the monophyly of the Nymphalis group (Nymphalis, Kaniska, Polygonia, and Inachis) within Nymphalini was also recovered in our study with high nodal supports (1.00 by BI and 100% by ML analyses), as has been supported in previous studies (Wahlberg and Zimmermann, 2000; Nylin et al., 2001; Wahlberg et al., 2005). The positions of Araschnia and Euphydryas each as the most basal lineage of Nymphlini and Melitaeini, respectively, were consistent with previous studies (Wahlberg and Zimmermann, 2000; Nylin et al., 2001; Wahlberg et al., 2005).
Nymphalinae is a well-supported monophyletic group that includes the four well-defined tribes: Nymphalini, Coeini, Melitaeini, and Kallimini (Freitas and Brown Jr, 2004; Wahlberg et al., 2003, 2005). In Melitaeini, both Wahlberg and Zimmermann (2000) using mtDNA sequences (COI and 16S rRNA), and Wahlberg et al. (2005) using combined mitochondrial and nuclear sequences, placed Euphydryas as the most basal lineage of Melitaeini, consistent with the present study. Nylin et al. (2001), Wahlberg et al. (2003), and Wahlberg et al. (2005) established the monophyly of the Nymphalini and the close relationships among Kaniska, Nymphalis, and Polygonia, as was verified in this study. However, the relationships of other genera within Nymphalini differ from the current study, possibly due to limited sampling in this study.
With respect to the tribe Argynnini in Heliconiinae, Simonsen (2006) subdivided this tribe into three subtribes based on 141 characters derived from wing and genitalia of males and females, with Euptoietina consisting of the genus Euptoieta, Yrameina consisting of the genera Yramea and Boloria, and Argynnina consisting of the genera Prokuekkenthaliella, Issoria, Brenthis, and Argynnis. By combining the morphological characters with new molecular data, Simonsen et al. (2006) subsequently suggested that the tribe Argynnini should be subdivided into four subtribes, placing the genus Boloria in a new subtribe Boloriina and proposed the subtribal relationships (Euptoietina + (Yrameina + (Boloriina + Argynnina))). In this regard, all genera employed in our study, except for Brenthis and Clossiana, belong to the subgenera of the genus Argynnis, while Clossiana belongs to a subgenus of the genus Boloria. Consistent with the data of Simonsen (2006) and Simonsen et al. (2006), our results indicated the three genera within the tribe Argynnini monophyletic groups and internal relationships among genera as ((Argynnis + Brenthis) + Boloria) in all analyses. Simonsen’s genus Argynnis was consistently split into two clades with high supports in all analyses in our study: the first one comprising one subgenera Fabriciana (represented by F. nerippe and F. adippe) and the other composed of the remaining Argynnis including subgenera Argynnis, Nephargynnis, Damora, Argyronome, Childrena, and Argyreus (Fig. 1). This result is also consistent with the data of Simonsen et al. (2006).
Within the subfamily Limenitidinae, the tribe Neptini composed of two genera (Neptis and Aldania) was recovered as a monophyletic group. Nevertheless, the genus Neptis represented by 10 species was revealed as paraphyletic with respect to the genus Aldania in all analyses (Fig. 1). Aldania raddei represented for the genus Aldania is somewhat larger in adult body size, although male genitalia characters are typical (Kim, personal communication). Nevertheless, the genus Aldania is known as an offshoot of Neptis from eastern Russia and China in temperate Palearctic region (Tuzov et al., 1997) and even has been considered synonymous to the genus Neptis in several studies (Yuan and Wang, 1994; Huang, 2003). Thus, taxonomic revision for the genus might be required after further critical analysis.
The tribe Limenitidini was sampled only for one genus of Limenitis (eight species) in our study. The monophyly of Limenitis was strongly supported in BI analyses (1.00), while moderately supported in ML analysis (79%) (Fig. 1). In contrast to our study, using the mitochondrial CytB gene, Wu et al. (2007) recovered as Limenitis paraphyletic. Zhang et al. (2011) also revealed Limenitis as paraphyletic by the ML method based on the COI gene alone. Although Zhang et al. (2011) reported a monophyletic Limenitis by the maximum parsimony method, the bootstrap value was very low (9%).
Overall, stable phylogenetic relationships among genera within tribes or subfamilies of Nymphalidae were obtained in all analyses, except for the conflict within Apaturinae that Apatura was placed as the sister to either Mimathyma or (Mimathyma + (Sephisa + (Hestina + Sasakia))). Nevertheless, the current study is the first extensive study for the Nymphalidae occurring in South Korea. Thus, the limitations of taxon sampling and sequence quantity were unavoidable, resulting in far less for completion compared with international criteria. Nevertheless, we expect the current work might be informative as the basal data for future work on the Nymphalidae occurring in South Korea.