다른 분율의 Cs이 포함된 Pt/CsxH3-xPW12O40 촉매를 제조하여 셀룰로우스의 폴리올로의 수소화분해반응를 수행하였다. 촉매의 비표면적과 Pt의 분산도는 Pt/CsxH3-xPW12O40 촉매의 Cs 함량이 증가함에 따라 증가하였다. 그러나 Pt/CsxH3-xPW12O40 촉매의 Cs의 함량이 변함에도 불구하고 비슷한 폴리올 수득률을 보였다. Pt/CsxH3-xPW12O40 촉매의 반응 활성은 Ni/W/SBA-15와 두 가지의 복합촉매(Pt/AC+ H3PW12O40과 Pt/AC+ Cs3.0PW12O40)와 비슷하였다. 이 반응을 진행하는 동안 Pt/CsxH3-xPW12O40) 촉매로부터 헤테로폴리 음이온이 침출되는 것을 확인하였다.
Biomass conversion into chemicals has attracted increasing attention in view of the raised concerns due to the limited reserves of fossil resources and the CO2 emissions responsible for the global warming. The recent high oil price is another driving force for the development of new processes in which non-petroleum-derived fuels and chemicals are produced from lignocellulose[1-3]. Because cellulose is the major component of non-edible lignocelluloses, it has the potential to become an alternative to fossil resources. On the other hand, cellulose is composed of strong β-1,4 glycosidic bonded glucose monomer, which led to difficult cellulose depolymerization.
Among the various chemical routes starting from cellulose, hydrogenolysis to polyols is considered to be a promising way to utilize cellulose as a chemical feedstock. Polyols (i.e., alcohols containing multiple hydroxyl groups) are important chemicals and also key intermediates for the manufacture of sweetener, foam insulation, fiber, adhesive, polyurethane, polyethylene terephthalate (PET), etc. With the aim to simplify the process, onepot transformations of cellulose into polyols have been investigated investi gated in the presence of hydrogen[4-18]. Since Fukuoka and Dhepe[4] first reported the conversion of cellulose into hexitols (e.g., sorbitol and manitol) over Pt/γ -Al2O3 catalyst, hydrogenation and/or hydrogenolysis of cellulose has been examined over Pt and/or Ru-based catalysts supported on various materials such as carbon black[5], activated carbon functionalized with sulfonated groups[6], carbon nanotubes[7,8], and H-ZSM-5[9]. With the help of reversible H+ ions formed from hot water above 473 K, Liu et al.[10] developed an efficient route for convertsion of cellulose into polyols using Ru/C. Transition metal-based catalysts such as Ni-W2C/AC[11], Ni-W/SBA-15[12], and Ni/ W/SiO2-Al2O3[13] have also been reported to be active for the production of ethylene glycol. Recently, combined systems containing both hydrolysis and hydrogenation catalysts such as heteropoly acids and Ru/C[14,15], Cs salts of heteropoly acids and Ru/C[16], mineral acid and Ru/C[17], and mineral acid and Ru/zeolite[18] were proposed for this reaction. Herein, cellulose is hydrolyzed over the acid catalyst, followed by metal-catalyzed hydrogenation into polyols. Geboers et al.[15] reported the remarkable potential of commercially available heteropoly acids in this reaction as compared to mineral acids. The heteropoly acids, having acidic and redox properties, are effective catalysts for several reactions. In these materials, the acid strength generally decreases in the following order: W6+ > Mo6+ > V5+ (for the polyatom) and P5+ > Si4+ ~ Ge4+ (for the heteroatom or the charge of polyanion).
Therefore, H3PW12O40 can be selected as a Keggin-type heteropoly acid with the highest acid strength. Since Keggin-type heteropoly acids are highly water soluble, a fraction of acidic protons should be replaced with other cations like Cs+ in order to make the material insoluble in an aqueous solution without changing the Keggin unit structure. This CsxH3-xPW12O40 can be used as a water-tolerant solid acid for environmentally friendly catalytic processes[19,20]. In this sense, CsxH3-xPW12O40 salts were found to be more active than the parent acid form and other known solid acid catalysts for liquidphase reactions such as the decomposition of cyclohexyl acetate, the alkylation of 1,3,5-trimethylbenzene, the acylation of aromatics, the hydrolysis of 2-methylphenyl acetate, and the hydrolysis of cellulose, as a result of their relatively high surface area and high surface protonic acidity[19-21]. The Pt catalyst supported on acidic cesium salts of H3PW12O40 (e.g., Pt/CsxH3-xPW12O40) can operate as a bifunctional catalyst as it contains both metallic Pt and CsxH3-xPW12O40 working as hydrogenation and acid sites, respectively.
In this work, Pt/CsxH3-xPW12O40 (x = 0.5-3) catalysts were prepared and applied to hydrogenolysis of cellulose in aqueous medium.
Acidic cesium salts, CsxH3-xPW12O40 (denoted as Csx hereafter), were prepared by adding the required amount of an aqueous cesium carbonate (0.47 M, Acros Organics) solution in a dropwise manner into an aqueous H3PW12O40 (0.75 M, Strem Chemicals, SBET = 2 m2/g) solution at room temperature upon vigorous stirring, as described by Okuhara et al.[22]. The Cs content, i.e., “x” in Csx, (e.g., 0.5, 1.0, 1.5, 2.0, 2.3, 2.5, 2.7, and 3.0) was adjusted by varying the amount of aqueous cesium carbonate solution added. Since the beginning of the addition of the cesium carbonate solution, a precipitate was formed resulting in a milky solution. The precipitate obtained was aged for 20 h at room temperature, followed by evaporation of water at 333 K. The precipitate was subsequently dried at 393 K overnight, and finally calcined under air at 573 K for 3 h.
All Pt/Csx catalysts were prepared using a wet impregnation method. The Csx catalysts, dried overnight at 393 K, were impregnated with an aqueous solution of tetraammineplatinum(II) nitrate (Pt(NH3)4(NO3)2, Aldrich), and the impregnated material was dried at 393 K overnight. The Pt content was 1 wt% in all cases, except for 0.1 wt% Pt/Cs2.5. All of the Csx-supported catalysts were calcined under air at 573 K and reduced in a H2 stream at 573 K for 1 h before the reaction. For comparison, 1 wt% Pt/activated carbon (Pt/AC, Aldrich) catalysts were purchased and utilized after reduction in a H2 stream at 573 K for 1 h before the reaction.
SBA-15 was synthesized using the method reported by Meynen et al[23]. Ni/W/SBA-15 catalyst was prepared using a sequential incipient wetness impregnation method. First, the support, dried at 393 K overnight, was impregnated with an aqueous solution of ammonium metatungstate hydrate[(NH4)6H2W12O40? xH2O, Aldrich] and the catalyst was dried at 393 K overnight. Second, this dried catalyst was impregnated again with an aqueous solution of nickel nitrate hexahydrate[Ni(NO3)2? 6H2O, Junsei Chemical Co.], and the catalyst was again dried at 393 K overnight. The Ni content was intended to be 5 wt%, and the content of W was intended to be 25 wt%. Ni/W/SBA- 15 was reduced in a H2 stream at 973 K for 1 h before the reaction.
2.2. Catalyst characterization
Thermogravimetric analysis (TGA) and drifferential scanning calorimetry (DSC) were conducted using a SIMULTANEOUS Thermal analyzer-Mass Spectrometer (STA 409pc + QMS 403c, NETZSCH) with the aim to measure the thermal change of the Csx catalysts from 303 to 1,173 K at a heating rate of 10 K/min in air.
The BET surface area was calculated based on the N2 adsorption data obtained using an ASAP 2020 unit (Micromeritics) at liquid N2 temperature. Before the measurement, the sample was degassed under vacuum for 6 h at 473 K. The surface area, external surface area, and pore size distribution curve were calculated by the BET, t-plot, and BJH desorption method, respectively.
The bulk crystalline structures of the Pt/Csx catalysts were determined using the X-ray diffraction (XRD) technique. The XRD patterns were obtained with a Rigaku D/MAC-III instrument operated at 50 kV and 30 mA using Cu Kα radiation. The assignment of the crystalline phases was carried out using the PCPDFWIN software (version 2.2) based on the ICDD database. The primary crystallite sizes of the catalysts were determined using the Scherrer’s equation .
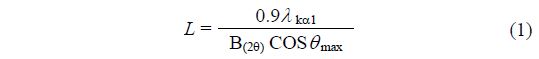
where L denotes the average crystalline size, 0.9 is the value when B(2θ) is the full width at half maximum (FWHM) of the peak broadening in radians, λ Kα1 is the wavelength of the X-ray radiation (0.15406 nm), and θmax is the angular position at the 26.1° corresponding to the peak maximum of Cs3.0.
The W, and Pt content for all of the prepared catalysts were
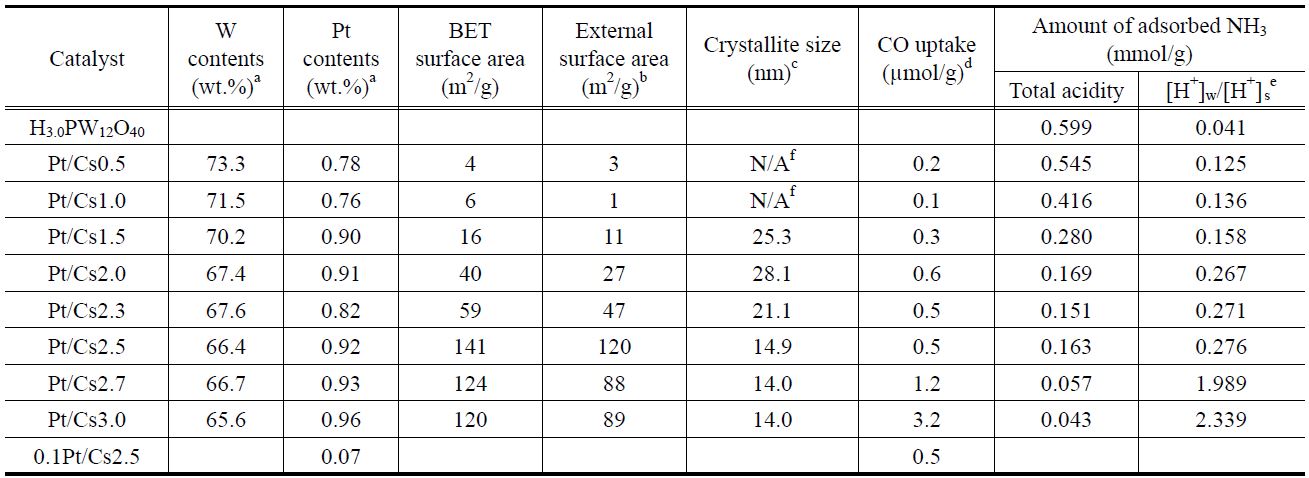
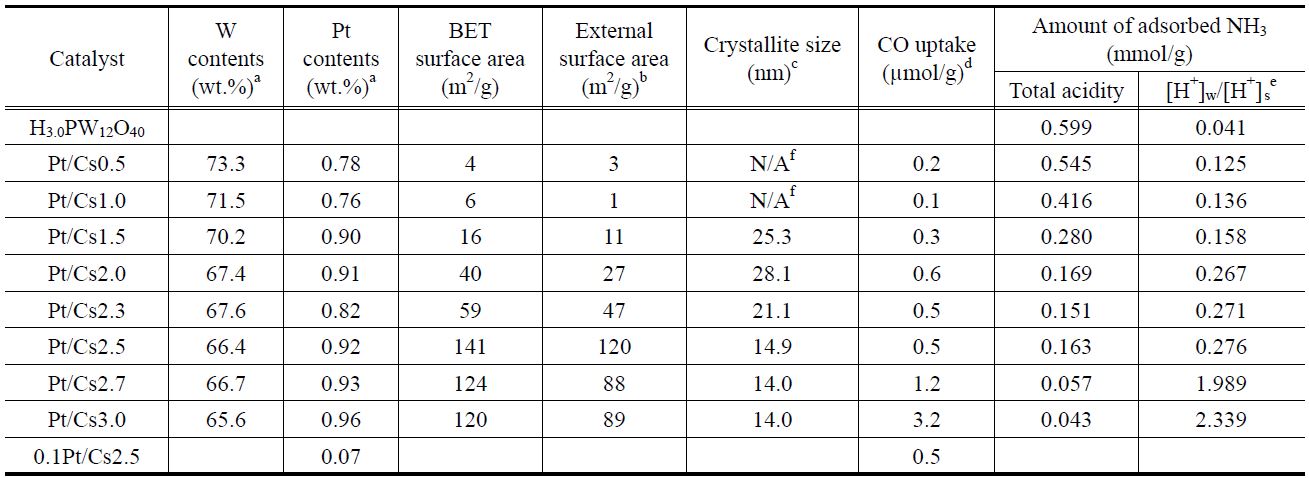
[Table 1.] Physicochemical properties of H3.0PW12O40, and Pt/CsxH3-xPW12O40
Physicochemical properties of H3.0PW12O40, and Pt/CsxH3-xPW12O40
calculated by inductively coupled plasma-atomic emission spectroscopy (ICP-AES, JY-70Plus, Jobin-Yvon), and the results are listed in Table 1.
CO chemisorption was carried in an AutoChem 2910 unit (Micromeritics) equipped with a thermal conductivity detector (TCD) to measure CO consumption. A quartz U-tube reactor was generally loaded with the sample, and the catalyst was pretreated by reduction in H2 at 573 K for 1 h, and subsequently cooled to room temperature. The CO chemisorption experiments were carried out at 300 K under a He stream (flow rate of 30 mL/min) by means of a pulsed-chemisorptions technique (500 μ L pulses) after removing any residual hydrogen in the line by flowing He at 300 K for 1 h.
The pulsed NH3 chemisorption experiments were carried out in a small fixed bed reactor connected to a gas chromatograph (HP5890) equipped with a TCD to measure the NH3 consumption. The tubular stainless-steel reactors were generally loaded with 0.50 g of the sample. All of the catalysts were pretreated in He at 573 K. The chemisorptions experiments were carried out at 573 K and 423 K, separately, using the pulsed-chemisorption technique (1 mL pulses). The amount of chemisorbed NH3 at 573 K and 423 K corresponded with the amount of strong acid sites and the amount of total acid sites, respectively. Therefore, the amount of weak acid sites can be derived by subtracting the amount of strong acid sites from the total amount of acid sites.
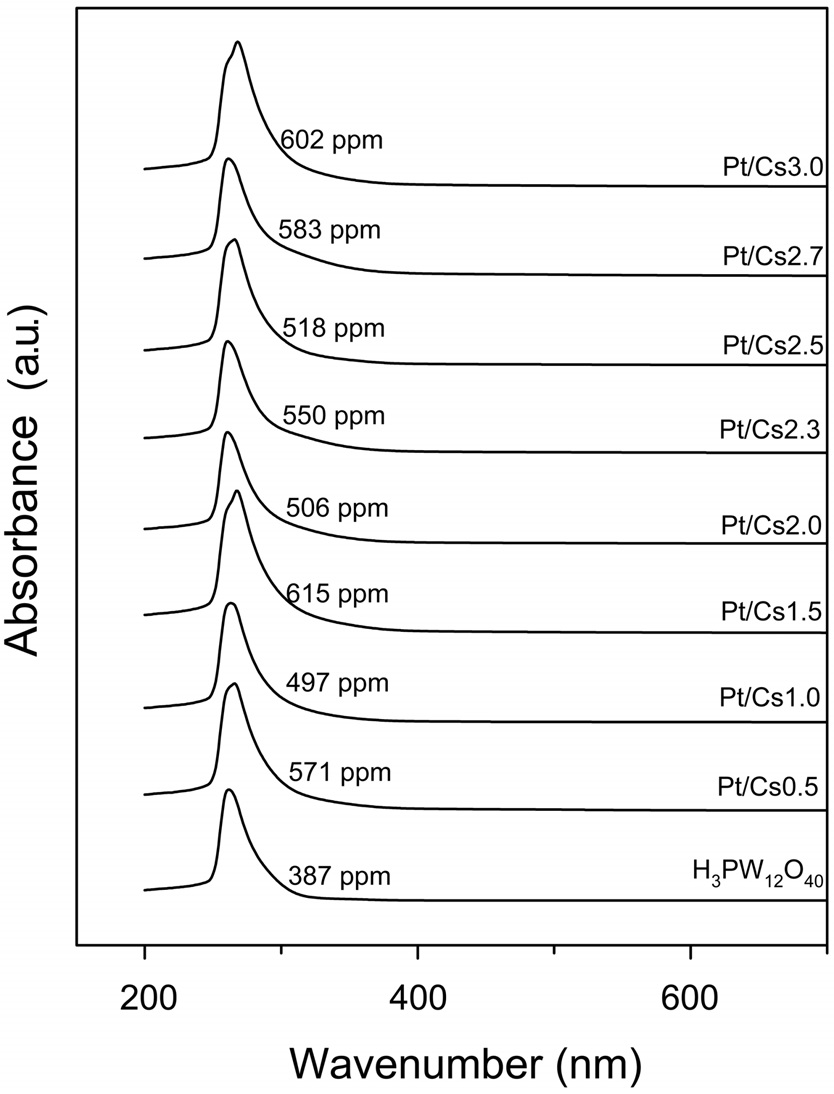
The concentration of heteropoly species present in the aqueous phase was measured by UV spectroscopy using a V-650 (JASCO) spectrometer. An adsorption band at around 260 nm characteristic of Keggin anions PW12O403-, was used for the analysis.
2.3. Catalytic performance tests
The relative crystallinity of cellulose (Aldrich, microcrystalline) was determined to be 84%[24]. Its direct conversion into polyols was carried out in a batch reactor. First, 500 mg of cellulose and 50 mg of the catalyst were mixed with 30 mL of deionized water, and the resulting mixture was introduced into an 85 mL stainless-steel autoclave. The reactor was then filled with H2, and the pressure was fixed at 6 MPa at ambient temperature. The reaction mixture was mixed well with the help of a magnetic stirrer (700 rpm). It took 1 h to reach the reaction temperature. Zero reaction time was taken as the time when the temperature reached the set temperature. After the reaction, the autoclave was cooled to room temperature within 0.5 h, and the solid residues were separated from the liquid products by centrifugation. The liquid product was filtered with a 0.2 μm membrane filter and analyzed with a high-performance liquid chromatograph (Waters, 1525 Binary HPLC Pumps) equipped with a refractometer detector (Waters).
With the aim to evaluate the cellulose conversion, the solid product was washed several times using deionized water and dried overnight at 353 K under vacuum. The cellulose convertsion and the product yield were calculated based on the following formulas:
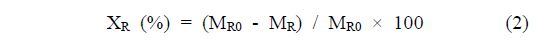
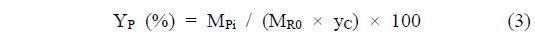
where XR is the cellulose conversion, MR0 is the weight of cellulose before the reaction, MR is the weight of cellulose after the reaction, YP is the yield for products, MPi is the weight of carbon in the products calculated considering carbon numbers, and yC is the weight fraction of carbon in cellulose determined using CHN analysis[24].
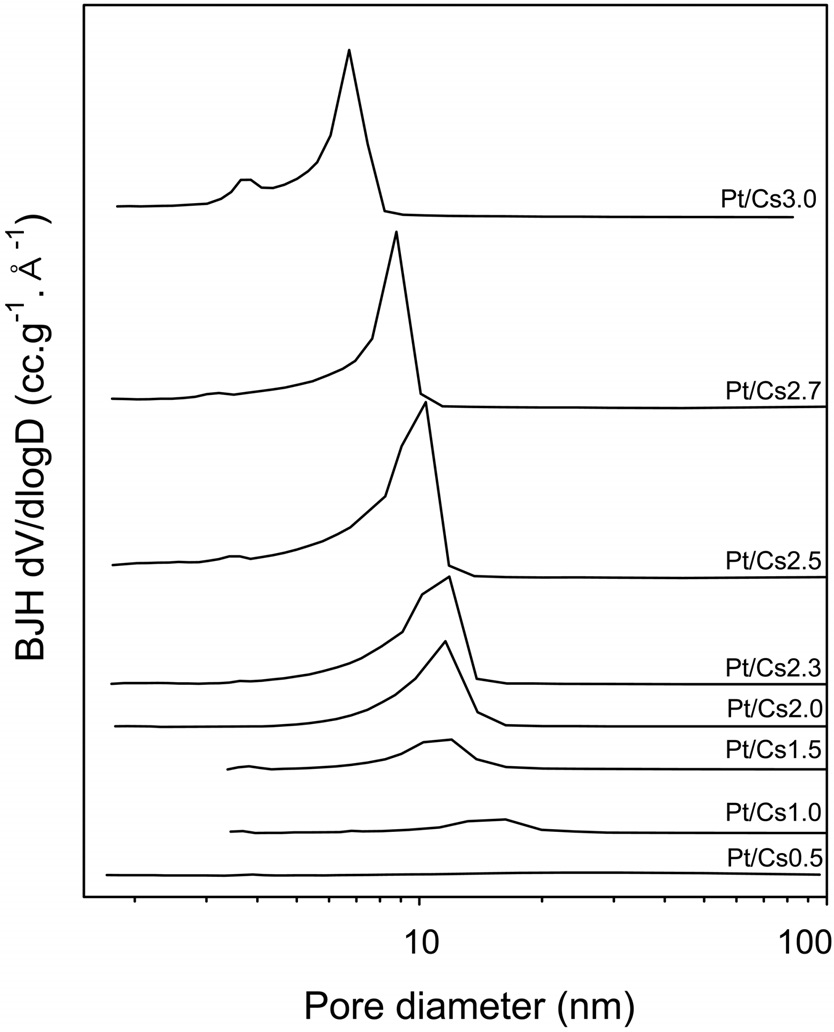
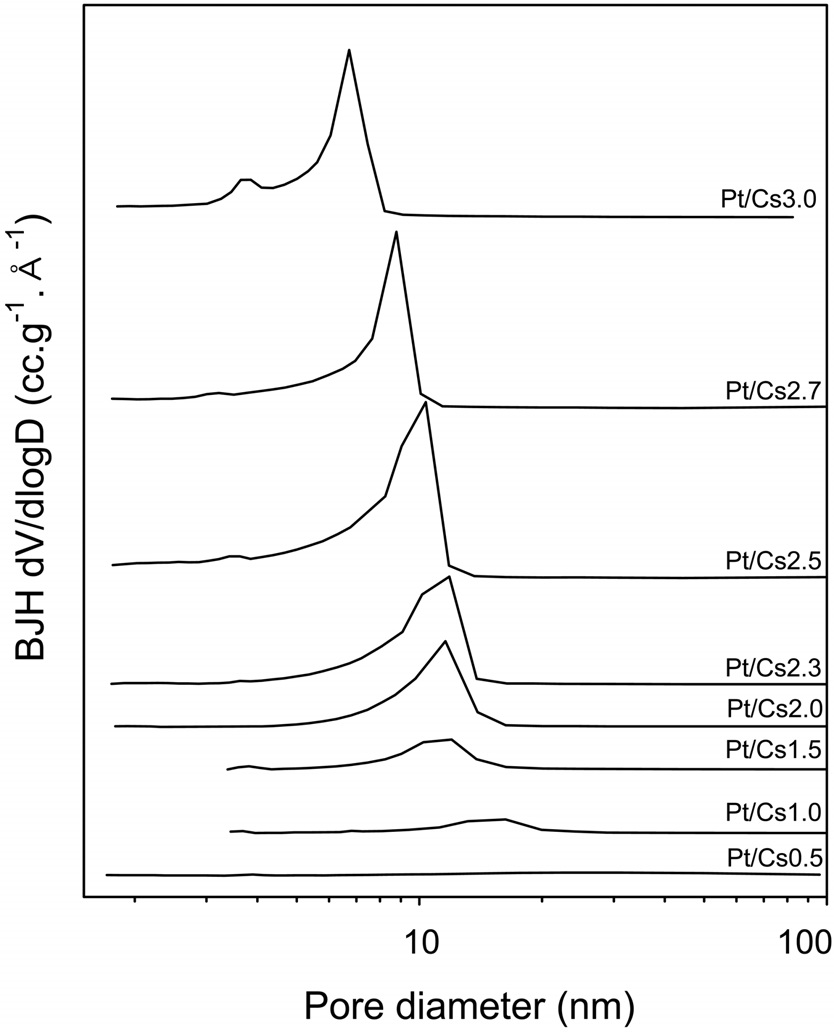
The physicochemical properties of all the catalysts under study were measured and listed in Table 1. It was confirmed that the W content in each catalyst measured with ICP analysis was in agreement with a theoretical value. Therefore, we can say that the intended metal content was loaded successfully. We prepared Csx following the method proposed by Okuhara et al.[22]. They proved that the ratios between Cs and polyanion of prepared catalysts were close to those of intended catalysts. A close correlation between the Cs content and the physical properties of Pt/Csx catalysts determined with N2 physisorption was found. Thus, both the BET surface area and the external surface area increased with increasing Cs content, reaching a maximum value for Pt/Cs2.5, and then decreasing for higher Cs contents. The pore size distribution for each Pt/Csx catalyst was obtained from the desorption branches of the N2 isotherms, as shown in Figure 1. The average pore diameter generally decreased with increasing Cs content for Pt/Csx catalysts. It has been reported that the mesopores are voids between the primary particle sizes (of about 10 nm) and that the micropores correspond to the spaces between the crystal planes formed by misfits[22]. The surface acid density for each catalyst was also measured by means of NH3 chemisorption (Table 1). As shown in Table 1, the total acidity decreased with the Cs content, and this can be explained in terms of a partial exchange of H+ with Cs+. The weak acid sites/strong acid sites ratio was also found to increase with Cs content, thereby implying that the stronger acid sites are preferentially exchanged with Cs+. The residual acidity of Cs3.0 is evidence for the fact that the ion exchange of Cs for protons of H3PW12O40 does not seem to be entirely quantitative. These incomplete ion-exchange phenomena are in line with the previous report that the completeness of the ionexchange process varied from approximately 5 to 70%, depending on both the solid phase and liquid phase cations for ion exchange of the ammonium, potassium, and cesium salts of 12-tungstophosphoric and 12-molybdophosphoric acids[24].
CO chemisorptions were also carried out for all of the Pt/Csx catalysts with the aim to determine the number of CO adsorption sites for each catalyst (Table 1). The amount of chemisorbed CO generally increased with Cs content. This implies that the Pt dispersion increases with Cs content. Na et al.[25] suggested that Pt in Pt-Csx worked as a site for hydrogenation/dehydrogenation and also it supplied hydrogen atoms to reactants.
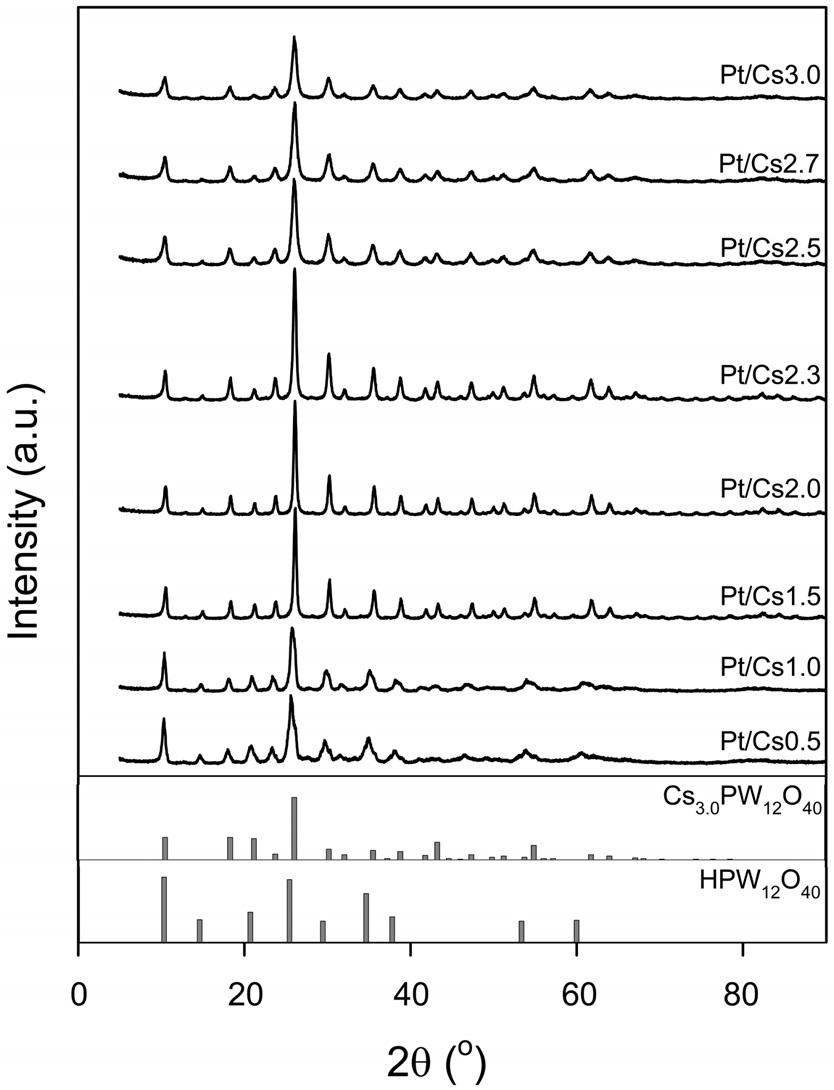
Figure 2 shows the X-ray diffraction patterns of the Pt/Csx
catalysts. XRD patterns produced by Cs3PW12O40 (JCPDS No. 50-1857) were found for all the catalysts under study. The Pt/Csx catalysts with low Cs content (e.g., Pt/Cs0.5 and Pt/Cs1.0) also showed XRD peaks assigned to H3PW12O40 (JCPDS No. 75-2125). Okuhara et al.[22] proposed a formation model of Csx in which Cs3.0 crystallites are first formed, and H3PW12O40 is subsequently adsorbed epitaxially on the surface of Cs3.0 during the titration and drying. In case of Csx catalysts with low Cs content (x < 2), ultrafine precipitate Cs3.0 particles are thickly covered with H3PW12O40, which is deposited upon evaporation of water, forming large aggregates[22]. Since the XRD peak intensity is closely related to the crystalline size, it can be said that the crystalline size of Cs3.0PW12O40 decreased with increasing Cs content, as indicated in Table 1. The crystalline sizes for x = 1.5-3.0 catalysts were in the range of 14.0-28.1 nm, and this crystalline size corresponds to the size of the primary particles. No XRD peaks due to Pt species can be observed because amorphous phases and/or crystallites with very low contents are invisible by the conventional XRD techniques.
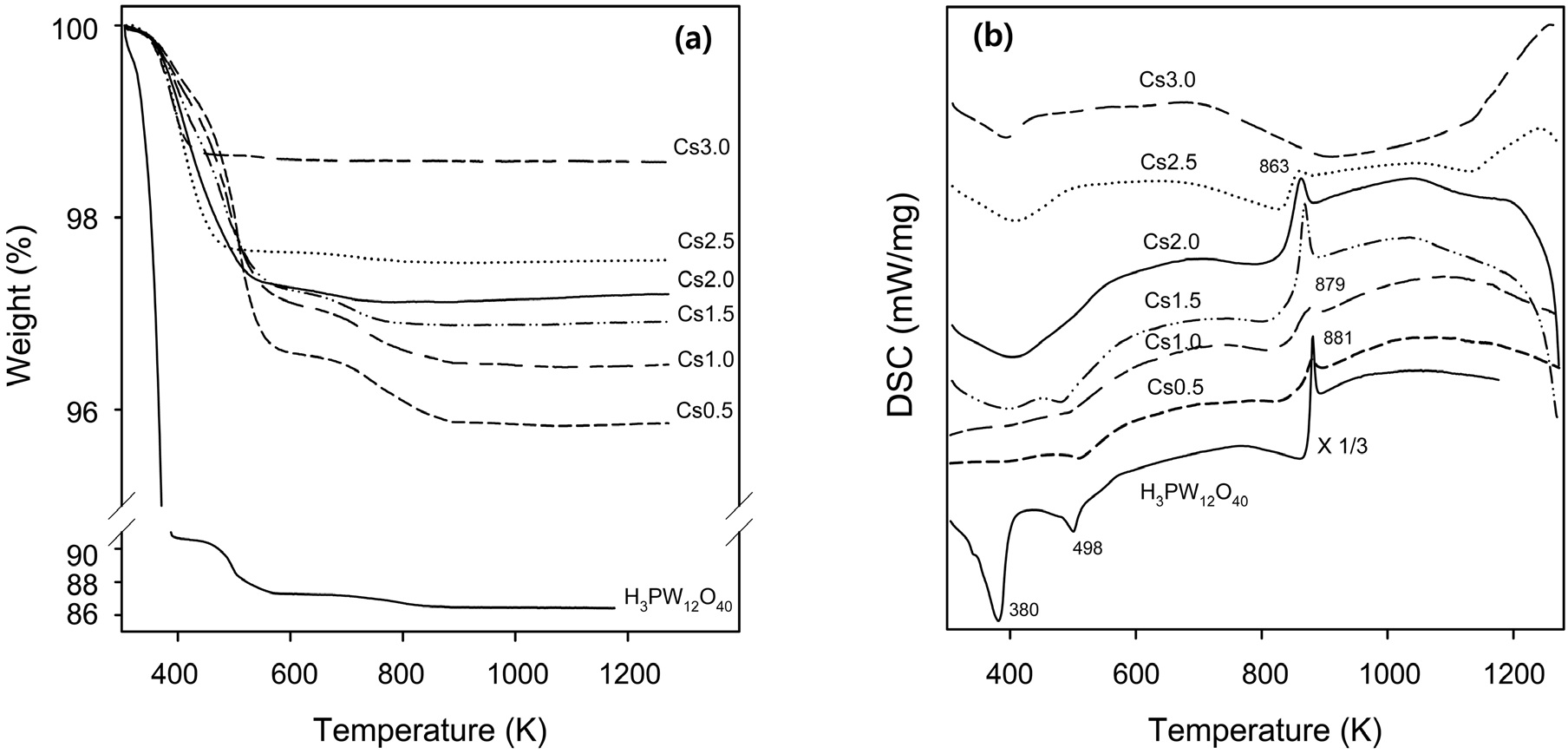
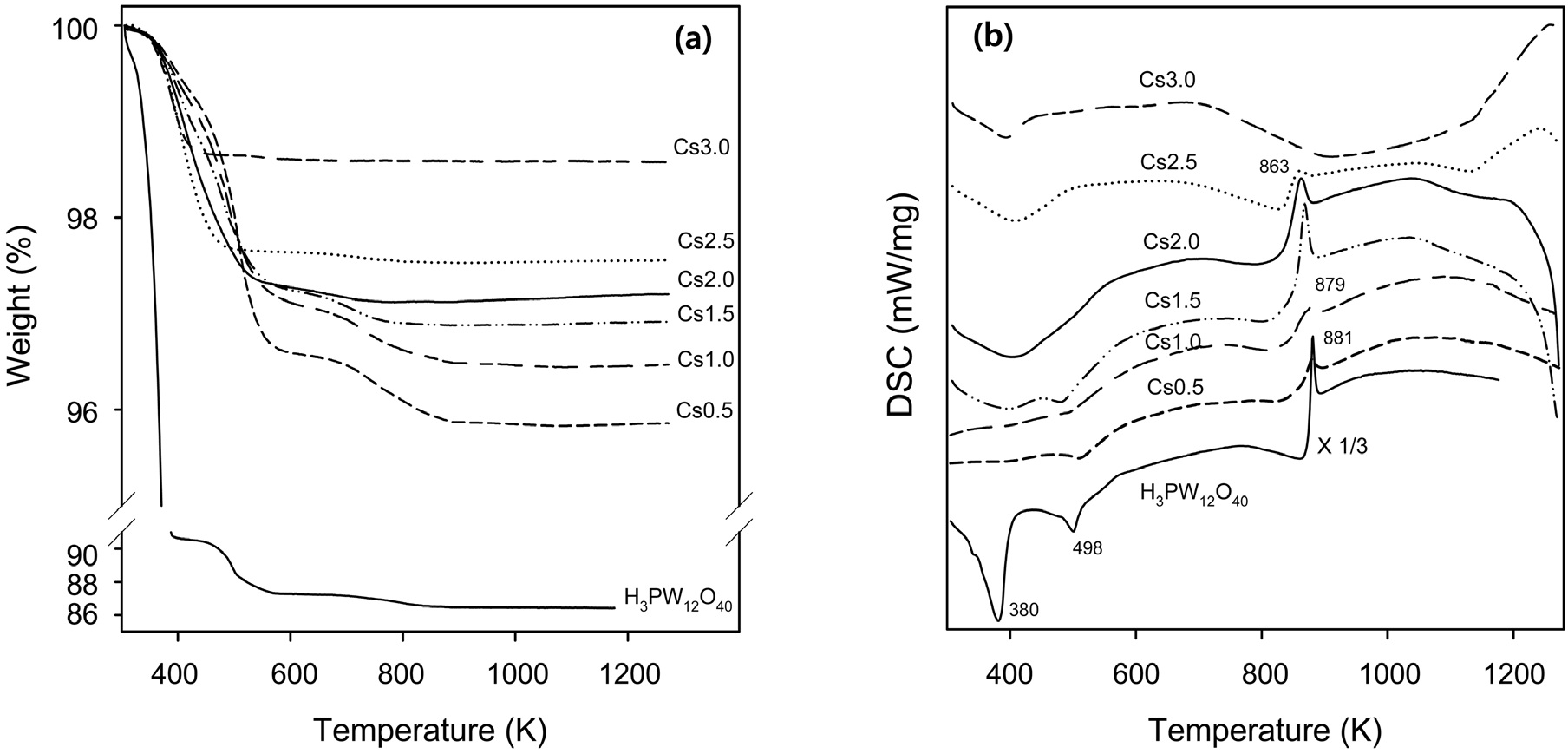
Thermogravimetric (TG) analysis was carried out for both H3PW12O40 and Csx supports, as shown in Figure 3(a). The main weight loss of H3PW12O40 was observed below 400 K. On the other hand, all of Csx supports showed the main weight loss below 600 K. An additional weight loss was found for H3PW12O40 below 600 K. In case of Cs0.5, Cs1.0, and Cs1.5, an additional weight loss was also observed below 880 K. These two weight-loss steps are related to two endothermic DSC peaks in the temperature region below 600 K (Figure 3(b)), indicating the release of physisorbed water and six molecules of combined water per Keggin unit, respectively[26]. The difference in the temperature can be explained as the removal of water in Csx
supports proceeds more slowly than that in H3PW12O40 because of the presence of ultramicropores in Csx supports[27]. The amount of removed water is dependent on both the Cs content and the preparation conditions. The amount of fractional weight loss was found to decrease with Cs content. The third exothermic DSC peak around 870 K with no weight change associated in TGA corresponds to the collapse of the Keggin heteropolyanion.
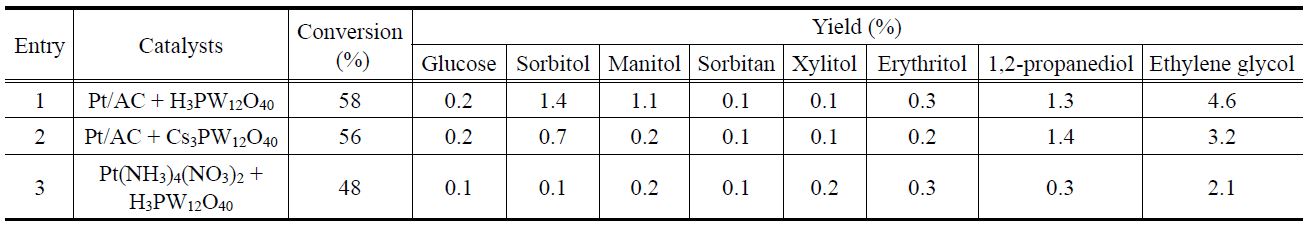
The direct hydrogenolysis of cellulose over the Pt/Csx catalysts was examined in the presence of H2, and results are listed in Table 2. The conversion of cellulose was 39% in the absence of any catalyst. This implies that in situ generated H+ ions in hot water as well as acid sites in the catalyst can play a role in the cleavage of the glycosidic linkages in cellulose. However the formation of polyol was no detected in the absence of a Pt catalyst.
Glucose and various polyols, including sorbitol, manitol, sorbitan, xylitol, erythritol, 1, 2-propanediol, and ethylene glycol (EG), were quantified. In all cases, the yield of glucose was lower than 1%, thereby implying that hydrogenation and/or hydrogenolysis of glucose is fast. In this reaction, ethylene glycol was the major product, and the yields of other polyols were lower than 3%. Chambon et al.[28] carried out cellulose conversion with Cs2.0 in the presence He obtaining carboxyl acid, gaseous product, and soluble oligomers/polymers as reaction products. Remarkably, these authors did not detect glucose and 5-hydroxymethyl furfural (HMF). The polyol yields obtained herein were lower than those reported in previous works as a much smaller amount of catalysts (cellulose/metal ratio of 1000 (w/w)) was utilized in this work compared to cellulose/metal ratios of 25-320 (w/w) reported elsewhere[4-8,14-18].
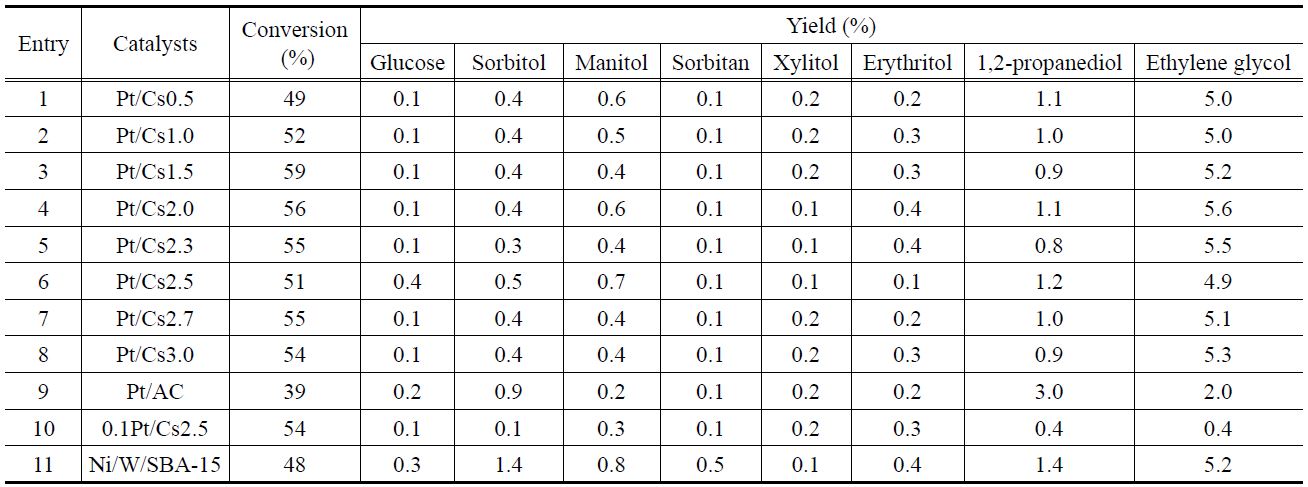
Cellulose conversions and yields of polyols by the transformation of cellulose into polyols over various catalystsa
The effect of the Cs content in the Pt/Csx catalysts on the cellulose conversion into polyols was examined in the presence of H2 (entries 1-8, Table 2). Although each Pt/Csx catalyst has a different surface acidity, Pt dispersion, and textural properties, no noticeable differences in the catalytic activity were found among them. Herein, the transformation of cellulose into polyols was carried out over Pt/Csx catalyst instead. Therefore, a balance between the acid function (for hydrolysis) and the metal function (for hydrogenolysis) is important to obtain a high yield of polyols, which implies that there might be an optimum composition for Pt/Csx catalysts. However, no correlation between the polyol yield and the catalyst composition was found. For comparison, a 5%Ni-25%W/SBA-15 catalyst, which was reported to show the high catalytic activity for this reaction[12], was also examined under the same reaction conditions. Compared with Pt/Csx catalysts, 5%Ni-25%W/SBA-15 showed slightly higher yield of sorbitol and manitol, although it exhibited similar yields of other polyols.
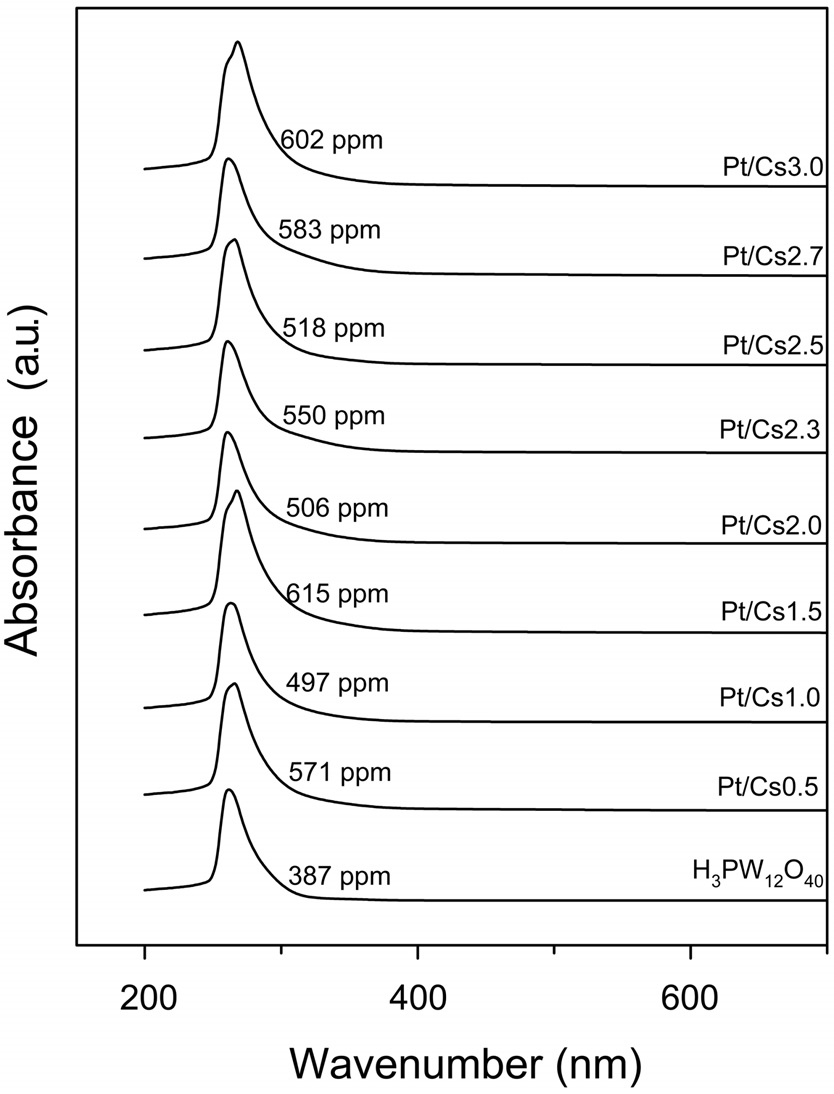
To resolve this inconsistency, the participation of homogenous catalysts leached from Pt/Csx was considered. UV spectra were obtained for the supernatant liquid after reaction in the presence of Pt/Csx catalysts. It was reported that Keggin-type anion, PW12O403-, showed two absorption bands near 200 and 260 nm [19,21]. In all cases, they exhibited an absorption band ascribed to the Keggin structure polyanions (Figure 4). The concentrations of W in the liquid phase after a reaction were determined to be 497-615 ppm, as shown in Figure 4. This implies that the fraction of W leached is 0.47, 0.42, 0.53, 0.45, 0.49, 0.47, 0.53, and 0.55 for Cs0.5, Cs1.0, Cs1.5, Cs2.0, Cs2.3, Cs2.5, Cs2.7, and Cs3.0, respectively. On the other hand, the Pt concentration in the liquid phase after reaction was below the detection limit of the ICP-mass (1 ppb).
To clarify the contributions of the homogeneous catalyst in this reaction, two different catalyst systems (e.g., Pt/AC + H3PW12O40 and Pt/AC + Cs3.0) were examined (entries 1 and 2, Table 3). It is worth mentioning that the amount of chemisorbed CO for Pt/AC were determined to be 24.0 μmol/gcat. No noticeable differences in the cellulose conversion and product distribution were found in these catalytic systems compared to Pt/Csx catalysts. However, when only Pt/AC was used (entry 9, Table 2), a smaller yield of EG was obtained compared to that obtained when Pt/AC + H3PW12O40 and Pt/AC + Cs3.0 were used. Therefore, it can be said that some Keggin structure polyanions were leached from Pt/Csx catalysts during the reaction, and that these polyanions play a role in this reaction.
In order to confirm that Keggin structure polyanions can be leached under hydrothermal conditions, Pt/Cs2.5 catalyst was exposed to hot water (463 K) for 12 h in the presence hydrogen. The supernatant liquid was subsequently analyzed resulting in a W detected concentration of 188 ppm, which corresponds to 16.0% of the original W content. It is worth mentioning that 47.0% of the original W content leached under the same reaction conditions in the presence of cellulose. This implies that leaching of polyanions from Pt/Csx catalysts can occur under hydrothermal reaction conditions. Furthermore, this leaching process might be accelerated in the presence of acids such as lactic acid, levulinic acid, and formic acid, which are formed from cellulose during the reaction[28]. In the case 5%Ni-25%W/ SBA-15 catalyst, 166 ppm of W was also detected in the supernatant after a reaction with this catalyst corresponding to 39.9% of the original W content.
0.1Pt/Cs2.5 catalyst, which has ten times lower Pt content as compared to Pt/Cs2.5 but shows a similar amount of chemisorbed CO as compared to Pt/Cs2.5, was also examined under the same reaction conditions. (entry 10, Table 2). Although it showed a similar cellulose conversion compared to other Pt/Csx catalysts, it exhibited much lower polyol yields. This implies that the surface structure of the catalyst might be altered during the reaction under aqueous phase conditions, and that embedded Pt can be utilized under reaction conditions. The very low amount of chemisorbed CO for Pt/Csx catalysts might be caused by the surface migration of Csx species onto the surface of Pt metal during the calcinations/reduction steps. The change of the catalyst surface can be inferred from the fact that some polyanions are leached during the reaction. XRD data for the solid residue after a reaction (data not shown) reveal that the XRD
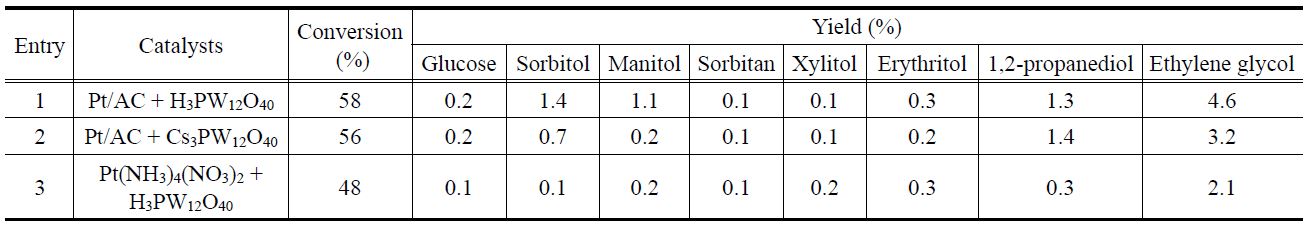
Conversion of cellulose and yields of polyols by the direct conversion of cellulose into polyols over combined catalytic systemsa
peaks corresponding to Cs3.0PW12O40 phase get sharpened and the weak XRD peak corresponding to Pt is observed. This implies that the crystallinity of Cs3.0PW12O40 phase increased and the metallic Pt was stabilized under reaction conditions. In order to find out the participation of the homogeneous Pt species, the Pt precursor, Pt(NH3)4(NO3)2, was also used as a catalyst with H3PW12O40 for this reaction (entry 3, Table 3). A much lower yield of EG was obtained over this catalytic system compared with Pt/Csx catalysts thereby implying that the homogeneous cationic Pt species are inferior to the Pt metal in Pt/Csx catalysts under these reaction conditions.
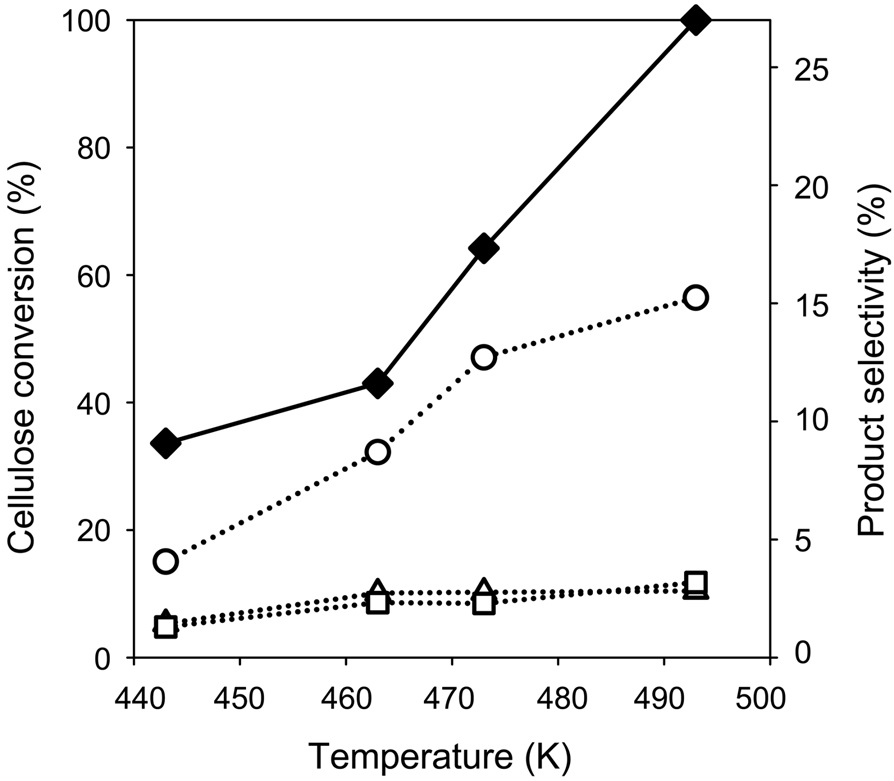
The effect of the reaction temperature on the conversion of cellulose into polyols was examined over Pt/Cs2.5, as shown in Figure 5. Both the cellulose conversion and EG yield increased with reaction temperature. The yields of 1,2-propanediol and hexitol were also found to be increase with reaction temperature.
The effect of Cs content in Pt/CsxH3-xPW12O40 on the aqueousphase hydrogenolysis of cellulose was examined. The BET surface area and the external surface area increased with increasing Cs content, reaching a maximum value for Pt/Cs2.5, and then decreasing for higher Cs contents. The total acidity decreased but the weak acid sites/strong acid sites ratio increased with the Cs content. Although the Pt dispersion increased with Cs content, the Pt dispersions for all Pt/Csx catalysts were poor because the some heteropoly acids blocked the Pt sites. No noticeable difference in the catalytic activity for this reaction was found among Pt/Csx catalysts. However, these catalysts showed the comparable catalytic activity with 5%Ni-25%W/SBA-15 under the same reaction conditions. Keggin structure polyanions were leached from Pt/Csx catalysts during the reaction.