상업적으로 이용 가능한 과산화초산을 산화제로 사용하고, 적은 양으로 매우 빠르게 산화시킬 수 있는 효율적인 철복합체 [((phen)2(H2O)FeIII)2(μ-O)](ClO4)4 촉매를 사용하여 -10 ℃에서 1,3-부타다이엔을 에폭시화하였다. 에폭시화반응에 대한 온도 (-10 ~ -40 ℃)와 시간의 효과에 관하여 연구하였다. 에폭시화반응은 -20 ℃에서 약 5분 내에 거의 완결될 정도로 빨랐으나, 그 이하의 온도에서는 느려졌다. 부타다이엔의 수율은 반응시간에 따라 증가하였으며, 부타다이엔 양이 증가하면 수율도 증가하는 경향을 보였다. 실험에서 얻은 부타다이엔모노에폭사이드의 최고 수율은 90%였다.
Epoxides are an important class of industrial chemicals that have been widely used as raw materials for production of epoxy resin, paints, surfactants, medicines and so on, or the efficient intermediates in organic syntheses[1-4]. Their selective synthesis is attractive to both academic research and industrial production [5-7]. Especially, the epoxidation of alkene is one of the main routes for the production of epoxides. BMO is one of the most important epoxides, because it is very versatile and can selectively react with nucleophiles at three sites on the carbon backbone[8]. Catalytic epoxidation-oxygenation of olefins to form cyclic epoxide groups is an important industrial process[5-7]. The epoxidation of 1,3-butadiene to BMO is an attractive commercial process because there are many metal complex catalysts for the epoxidation. Besides, 1,3-butadiene is cheap and easily available from industry. BMO is readily used to prepare vinylethylene carbonate (VEC), which can largely improve the performance of the secondary battery as an additive[9]. During the synthesis of VEC, carbon dioxide reacts with BMO and is consumed. Therefore, BMO can be considered as a clean material to be used for a clean process.
Although a number of epoxidation processes that use various catalysts and oxidants have been developed, a chlorine-based noncatalytic process (the chlorohydrin process) and catalytic processes based on expensive oxidants are still used extensively[10]. In contrast to such classical “nongreen” processes, H2O2-based catalytic epoxidation has some advantages because it makes environmentally benign H2O as a sole by-product and because it is rather inexpensive compared with organic peroxides and peracids[11]. Transition metal compounds such as metalloporphyrin[12], titanosilicates[13], methyltrioxorhenium[14], tungsten compounds[15-18], polyoxometalates[19,20], and manganese complexes[21] have been employed as a catalyst for H2O2-based epoxidation, either homogeneous or heterogeneous. Unfortunately, these systems have disadvantages, such as low efficiency of H2O2 utilization and low selectivity to epoxides (or low stereospecificity)[12-20], limited number of applicable olefins[21], and high reaction temperatures[15-17]. Therefore, effective catalysts of H2O2-based catalytic epoxidation of a wide range of olefins at ambient temperature have been sought.
Uncatalyzed epoxidation of alkenes with peroxycarboxylic acids has been commercially employed to epoxidize internal alkenes of fatty acids. Peracetic acid can epoxidize electron deficient terminal alkenes even at high reaction temperatures and with extended reaction times that may significantly reduce the selectivity[22]. However, metal-catalyzed reactions can enhance the reaction rates and lower the reaction temperatures. Some chiral[MnIII(salen)]1+ complexes were reported to efficiently epoxidize aromatic or substituted alkenes with high yields using peroxycarboxylic acids at -80 ℃, where salen is
Debois et al.[29] showed that a cationic complex, [MnII((
In this paper, we synthesized BMO from 1,3-butadiene using Debois et al.’s iron complex as a catalyst and peracetic acid as an oxidant. The main focus of this study is to investigate the effect of temperature and reaction time on the epoxidation of 1,3-butadiene.
1,3-Butadiene (99.9%) was kindly obtained from SK Energy. FeIII(ClO4)3 hydrate (chloride < 0.005%), 1,10-phenanthroline (99 + %), 32% peracetic acid, acetonitrile (99%), potassium carbonate (≥ 99%) and diethyl ether (99.5%) were purchased from Aldrich and were used as received.
2.2. Synthesis of Catalyst [((phen)2(H2O)FeIII)2(μ-O)] (ClO4)4 Stock Solution
FeIII(ClO4)3 hydrate (0.197 g, 0.56 mmol) was dissolved in 0.5 mL water placed in a 20 mL vial. Phenanthroline of 0.2 g (1.1 mmol) was dissolved in 4.5 mL of acetonitrile and the resulting solution was added to the vial. The solution with a green-brown color was stirred for 5 min at room temperature. This stock solution of 0.055 M was stored at -15 ℃ to prevent decomposition of the complex.
2.3. Epoxidation of 1,3-Butadiene
A solution of acetonitrile containing 1,3-butadiene (3.5 mmol) and 32% peracetic acid (7 mmol) were taken in a 25 mL Erlenmeyer flask was cooled to the required temperature. To this solution, a 0.055 M solution of iron complex catalyst (8.75 × 10-3 mmol, 0.25 mol%) was added over the course of 2 min while stirring. The reaction was stirred continuously for the required time and quenched by adding diethyl ether (18 mL). The reaction flask was removed from the dry ice bath and 1 g of K2CO3 in 5 mL H2O was added. The solution was extracted with ether (3 × 25 mL). The product yield was calculated by using gas chromatography (GC) with decane as an internal standard.
The products were characterized by a nuclear magnetic resonance (NMR) spectrometer (Bruker DPX, 300 MHz). Chemical shifts (δ) are reported in ppm downfield from external SiMe4 (solvent: CDCl3). Quantitative analysis of the products was performed by gas chromatography (GC) (Donam Systems Inc. DS- 6200) equipped with a DB-5 column (L: 30 m, I.D.: 0.53 mm). The injector and FID detector temperatures were set to 200 and 250 ℃, respectively. Helium was used as the carrier gas. The column oven temperature began at 30 ℃, kept 5 min, raised to 200 ℃ at a rate of 10 ℃/min and kept for 10 min at the final temperature.
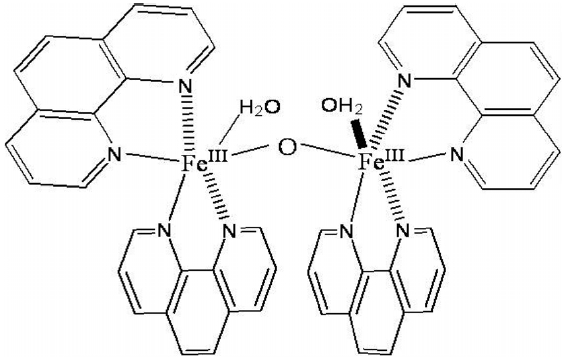
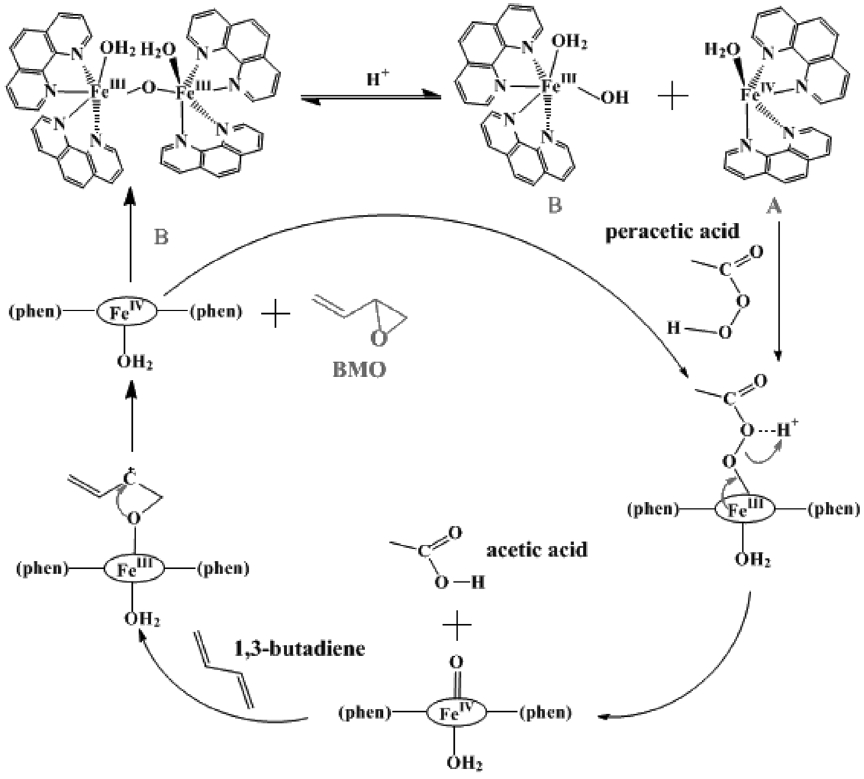
Epoxidation of 1,3-butadiene was performed to synthesize BMO using an iron complex as a catalyst and peracetic acid as an oxidant (Scheme 11). Catalyst iron complex contains two phenanthroline ligands ligate iron in cis coordination. The ferric species of catalyst prefer to dimerize in the presence of water, leading to a common structure, μ-oxo dimer such as [((phen)2(H2O)FeIII)2 (μ-O)](ClO4)4[31]. The bridging oxide ligand and water ligands occupy the adjacent cis sites of each iron. Due to the oxidative robust nature of phenanthroline, its metal complexes are great interest in the oxidation catalysis. Structure of the catalyst is shown
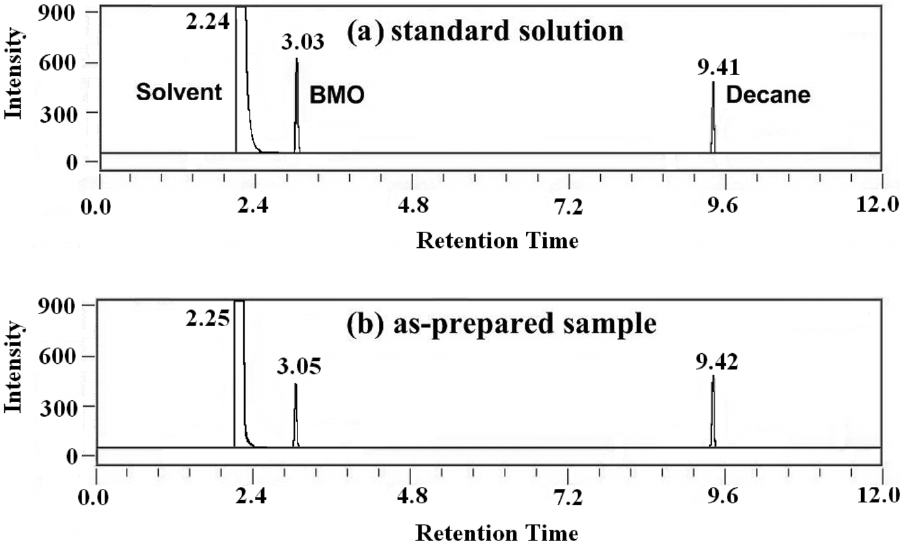
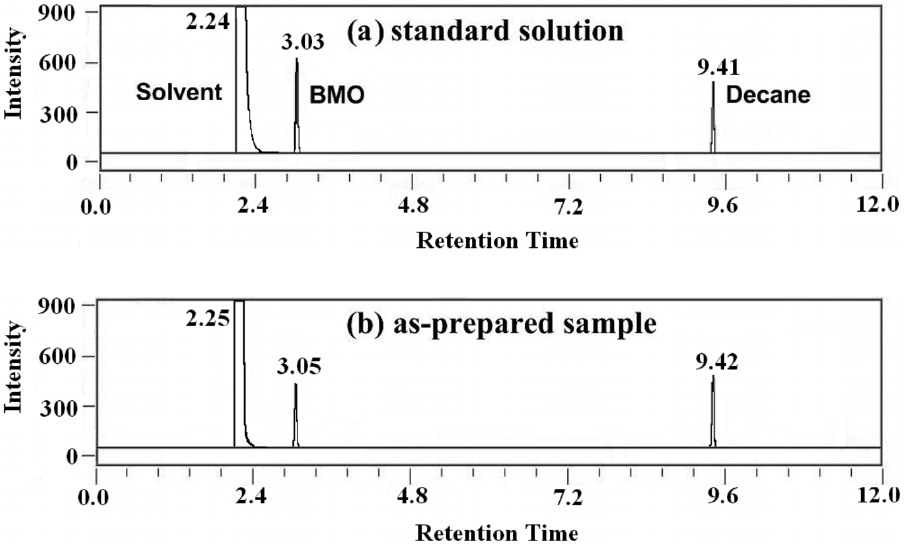
in Figure 1. When the catalyst is combined with peracetic acid, effective epoxidation occurs. Low catalyst loading of 0.25 mol% is sufficient to carry out the reaction. Slow addition of catalyst solution over 2 min to an efficiently stirred solution generally ensures efficient heat dissipation. The reaction was stirred continuously for the required time and quenched by adding diethyl ether. K2CO3 solution was added to neutralize acetic acid formed during the reaction. From the resulting solution, the product BMO was extracted using diethyl ether. The product yield was measured by using GC with decane as an internal standard. Figure 2 shows the gas chromatogram of standard (as received) and assynthesized BMO. From the GC interpretation (based on peak area), product yield was calculated.
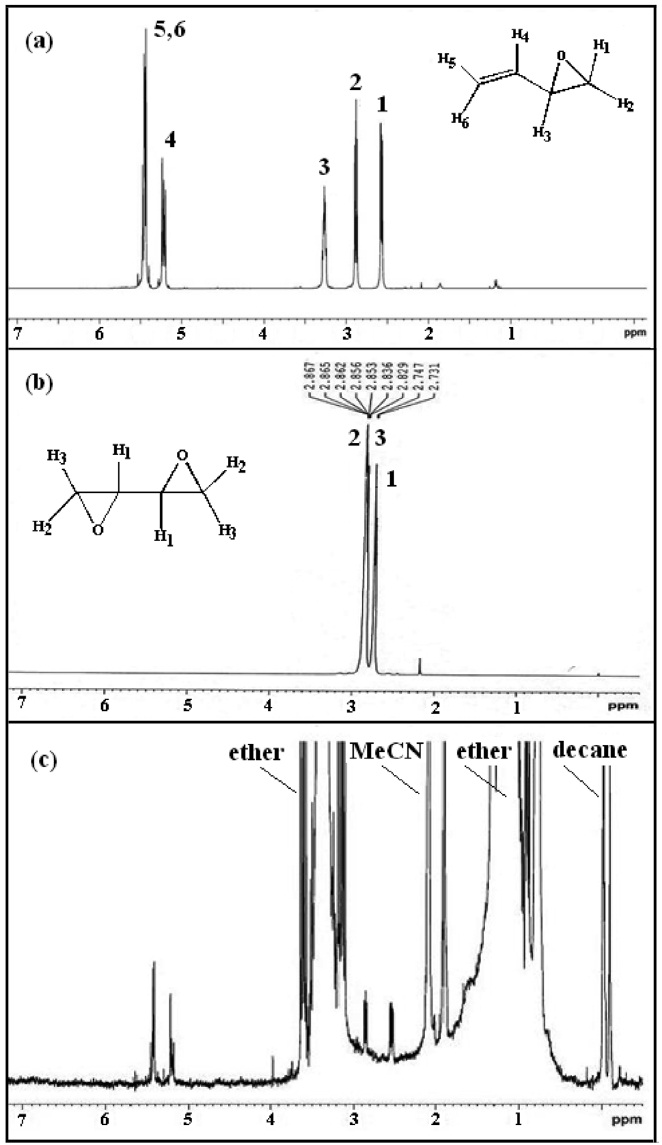
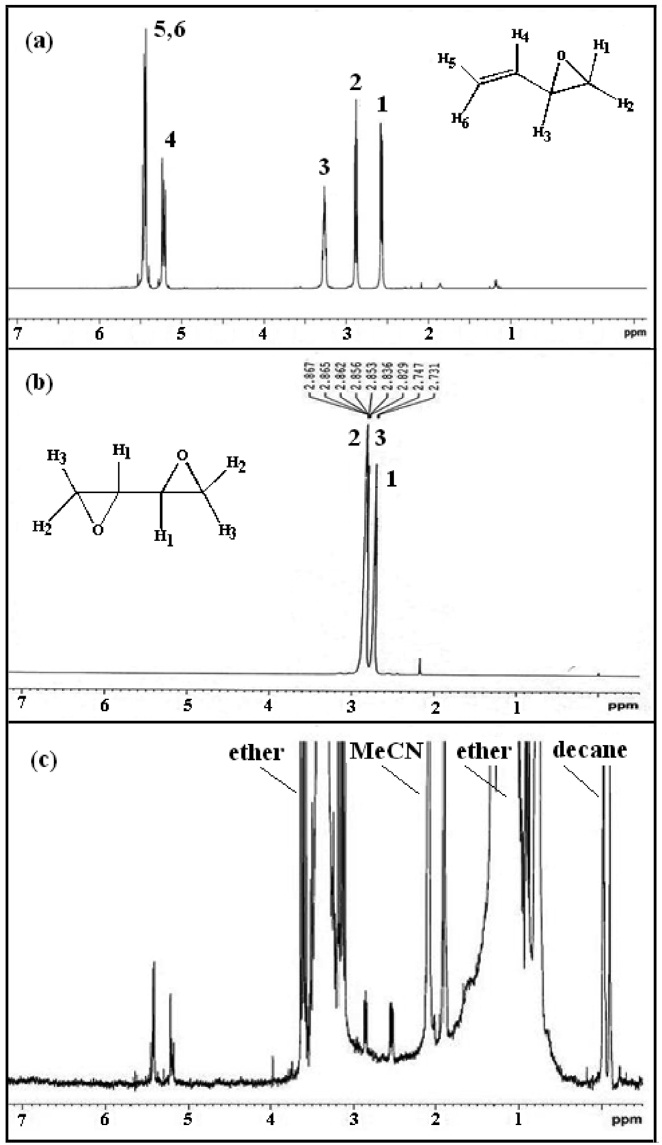
The structure of product BMO was confirmed by H-NMR shown in Figure 3. The chemical shift at δ = 2.60 and 2.89 ppm is associated with the methylene (= CH2) proton adjacent to the epoxide. The chemical shift at δ = 3.25 ppm attributes to the methine proton (CH) adjacent to the epoxide. The chemical shift in the region of 5.0~5.6 ppm corresponds to the methine proton in ethylene confirms the formation of BMO (Figure 3(a) and (c))
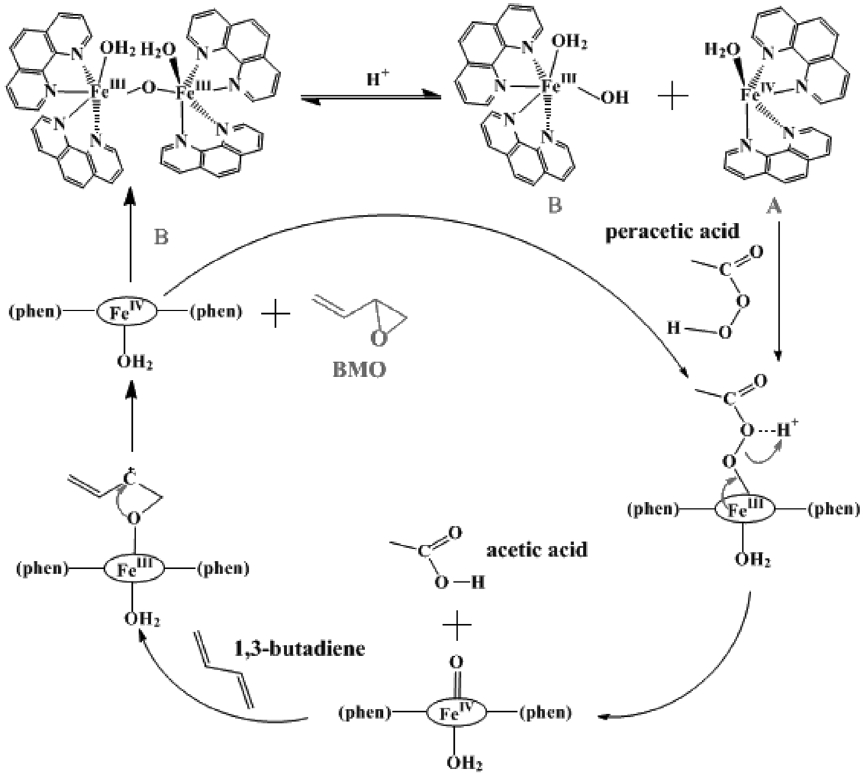
because all three chemical shifts for the protons in butadiene diepoxide are in the range of 2.5~3.0 ppm (Figure 3(b)). For butadiene, a C = C moiety with higher electron density was epoxidized regioselectively to give the corresponding monoepoxide without the successive epoxidation of the other C = C fragment (ie. no diepoxide is formed). The proposed mechanism[32,33] for the synthesis of BMO using oxidant peracetic acid and catalyst iron complex is shown in Scheme 21.
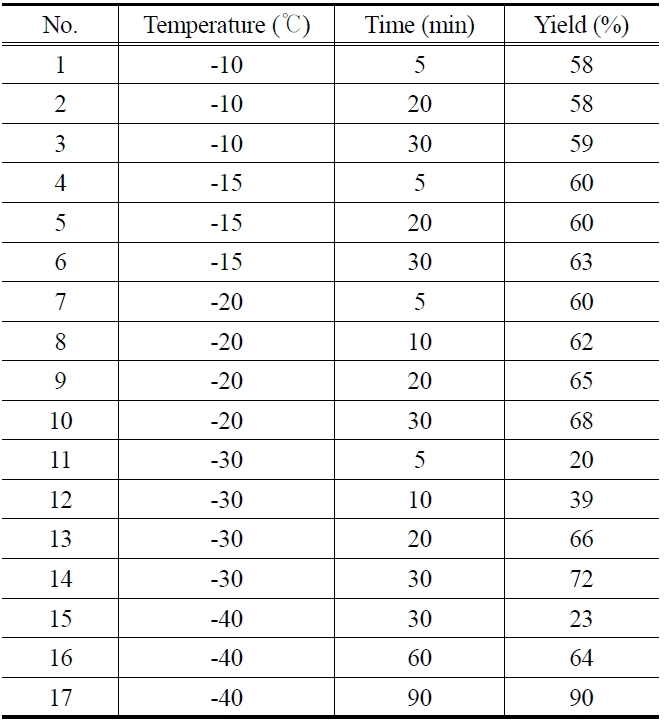
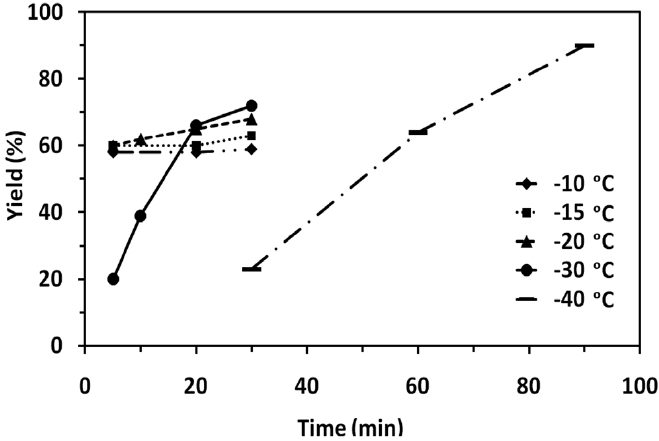
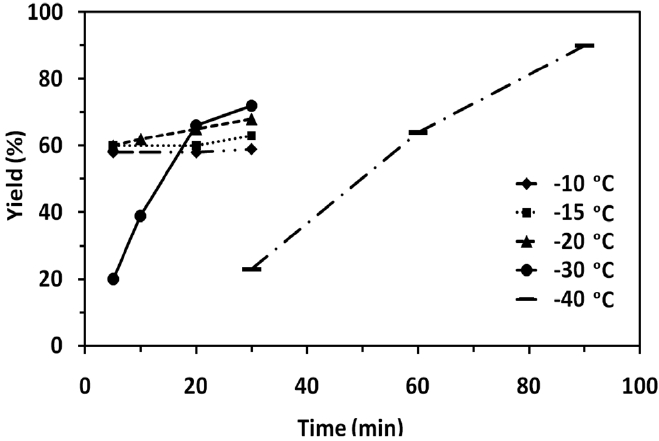
Table 1 shows the experimental data for the epoxidation of butadiene at different temperature and reaction time. In all the experiments, concentration of butadiene (3.5 mmol), peracetic acid (7 mmol) and catalyst (0.25% of 0.055 M solution) were kept constant. At reaction temperatures such as -10, -15 and -20 ℃, the epoxidation reaction was very fast and almost completed within 5 min (Table 1, entries 1, 4, and 7). The product yield was found to be 58, 60 and 60% respectively. There is no appreciable increase in the product yield when we further increased the reaction time to 20 and 30 min (Table 1, entries 2, 3, 5, 6, 9 and 10). When the reaction carried out at -30 or -40 ℃, the product yield increased with increasing the reaction time. At -40 ℃, the product yield increased from 23% to 90% (the best yield in this study) with increasing the reaction time from 30 to 90 min (Table 1, entries 15, 16, and 17). The effect of temperature and reaction time to the product yield is shown in Figure 4. This may be attributed to the fact that the rate of reaction becomes slower at -40 ℃.
[Table 1.] Epoxidation of 1,3-butadiene (0.3 ml) at different temperature and reaction time
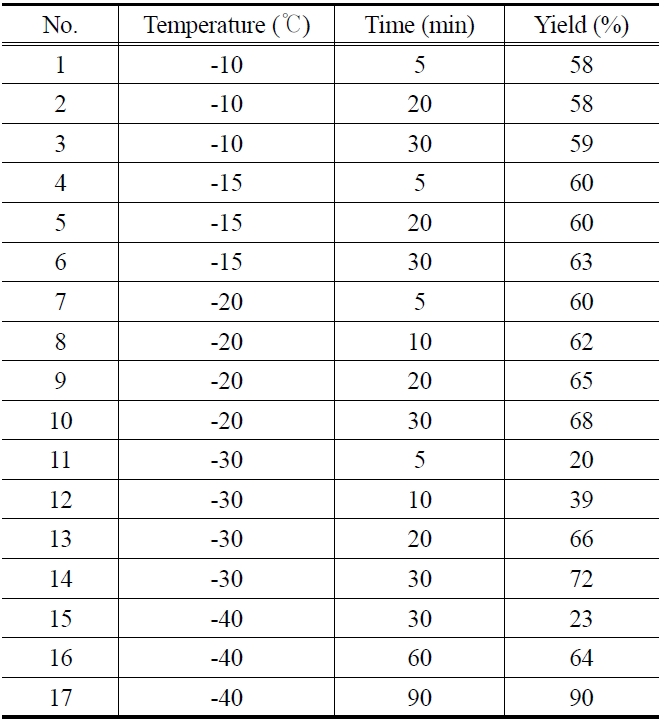
Epoxidation of 1,3-butadiene (0.3 ml) at different temperature and reaction time
There are several factors that can affect the product yield, including highly volatile nature of 1,3-butadiene, loss of BMO during the purification process and decomposition of BMO. In the purification of BMO from the reaction mixture, K2CO3 so
lution was used to neutralize the acetic acid formed in the reaction and diethyl ether was used (several times) to extract the BMO. During this process, some amount of BMO must have been lost by vaporization. Therefore, it is clear that the loss of volatile 1,3- butadiene by vaporization was minimal at -40 ℃, leading to the highest yield of 90%. Since, the epoxidation of butadiene is both exothermic and fast; large-scale reactions need to be performed in high surface to volume flasks (e.g. Erlenmeyer flasks) with external cooling to assure appropriate heat dissipation. The efficiency of both catalyst preparation and the epoxidation reaction itself provide significant advantages over other epoxidation procedures for termination olefins.
In summary, we have demonstrated the detailed investigation on the epoxidation of 1,3-butadiene at different temperatures (-10 to -40 ℃) and at different reaction time using iron complex [((phen)2(H2O)FeIII)2(μ-O)](ClO4)4 as a catalyst and peracetic acid as an oxidant. At temperature -10, -15 and -20 ℃, the epoxidation rate was very fast but the yields were not high due to the loss of considerable amount of highly volatile 1,3-butadiene. The highest yield of 90% was obtained at the reaction temperature -40 ℃. Further efforts are still in progress to tune the experimental conditions as well as to find new catalysts that can selectively epoxidize terminal olefins with a high conversion.


![Chemical structure of the catalyst[31].](http://oak.go.kr/repository/journal/11703/CJGSB2_2012_v18n1_51_f001.jpg)