산업 발달로 화석 연료 사용이 급증하고 이에 따른 지구 온난화 문제와 자원 고갈 문제가 대두되어 지속 성장을 위협하고 있다. 따라서 지속 성장을 위해서 두 문제를 모두 해결하여야 한다. 현재 이산화탄소의 처리 방법으로 인식되고 있는 이산화탄소 포집 및 저장 기술(carbon capture and sequestration, CCS)의 환경 논란으로 인해 사후 처리 기술의 필요성이 커지고 있다. 이 에 해결책중 하나로 부각되고 있는 이산화탄소 포집 및 재활용 기술(carbon capture and utilization, CCU)에 대해서 알아보았다. 이산화탄소 전환 기술은 이산화탄소 배출량 감소에 따른 지구 온난화 문제의 해결 뿐 아니라 탄소원의 재활용이란 측면에서 자원고갈 문제의 해결책으로 제시될 수 있겠다. 이산화탄소 전환 기술은 기상 전환과 액상 전환으로 나눌 수 있으며 기상 전환의 경우 필요 에너지 공급원과 온화한 반응조건에서 전환이 이뤄져야 하고 저에너지 소비 생성물 분리 정제 기술의 개발이 필요하다. 액상 전환의 경우, 반응 속도를 높일 수 있는 촉매 및 광감응제 개발과 함께 촉매, 빛, 전기의 혼성 시스템의 개발이 요구되어진다. 이산화탄소 전환 기술은 신재생 에너지 및 바이오산업의 경쟁력 향상을 위한 연결 기술로 그 가치가 매우 크다.
이산화탄소는 현 문명 세계를 유지하는 온실 역할을 수행하여 왔다. 그러나 인구 증가와 산업발전에 따른 에너지 소비의 급증은 유례없는 이산화탄소의 증가를 가져와 대기 중 이산화탄소 농도가 400 ppm를 넘어 설 것으로 보고 있다. 이는 지구 온난화 현상을 가져와 빙하 감소와 해수면 증가, 그리고 폭우 또는 가뭄 등 다양한 대형 자연재해가 일어나고 있으며 이는 평형점을 찾기 위한 현상으로 보고 있다. 그러나 임계점을 넘어서는 현상이 일어나면 다시 되돌리기는 불가능한 상황이 될 것이다. 이는 북극 빙하의 감소는 점차 가속되어가고 있는 것으로 확인할 수 있다.
유럽 연합 등 세계는 이산화탄소 농도를 500 ppm에서 안정시키고자 노력하고 있다. 이산화탄소를 저감하는 방법으로 에너지 효율화, 대체 에너지 개발 등 다방면의 노력을 경주하여 왔으나 이산화탄소의 증가는 계속되고 있다. 이는 신흥국의 발전과 인구의 증가에 따라서 에너지의 소비는 계속 증가되고 있기 때문이다. 에너지 소비 증가에 따라서 화석 연료의 사용은 향후 몇 십 년 아닌 그 이상 사용될 것으로 보이고 있다. 이는 태양전지, 풍력 등 신재생에너지로 화석 연료를 대체 할 수 없기 때문이다. 이는 이들 신재생에너지가 에너지 삼화음 (三和音, 밀도, 저장, 이송) 측면에서 기존 화석 연료를 대체 할 수 없기 때문이다. 따라서 발생되는 이산화탄소에 대한 사후 처리 기술이 필요로 하고 있으나 효과적인 방법이 제시되지 못하고 있다. 많은 환경론자들의 문제제기에도 불구하고 저장 기술이 가장 현실적인 사후기술로 보아 이들 처리 기술의 청정개발체제(clean development mechanism, CDM)사업을 인정하기에 이르고 있으나 저장 장소 제한 및 선정에 어려움이 있을 것으로 예견되고 있다. 탄소의 처리 기술은 자연계 이산화탄소 순환을 통해 탄소 사이클을 통하여 탄소(이산화탄소)를 처리하는 것이 보다 효과적이라 생각한다.
국제 기후 변화 협약 가입에 따른 온실가스 저감 목표 할당 제와 이에 따른 벌금 등 강제적인 요소가 도입되면서 최근 들어 전 세계적으로 녹색기술 산업에 대한 관심이 고조되고 이를 실현하기 위한 연구개발이 활발히 수행되고 있다. 지금까지 산업공정에서 대량 배출되는 대표적 온실가스인 이산화탄소를 모아 폐기물처럼 지하나 해저에 매립/저장하는 CCS에 주로 초점을 맞추어 연구가 진행되어 왔으나, 최근 들어 이 이산화탄소를 자원화하는 CCU 기술과 관련한 연구가 새로운 관심을 받고 있다. CCU 기술은 이산화탄소를 단순히 버려지는 물질이 아닌 유용한 자원으로 활용하여 부가가치가 높은 다 른 탄소화합물(value-added chemicals)로 전환하는 연구로써, 온실가스의 감축을 통해 환경 문제를 해결함과 동시에, 대기 중에 풍부하게 존재하는 이산화탄소를 탄소원으로 사용하고 또한 사용 후에는 다시 이산화탄소의 형태로 대기 중으로 배출 되므로 지속가능한 탄소원의 재활용이란 측면에서도 그 의미를 찾을 수 있다. 아울러 전환 후 고부가가치 탄소화합물의 생성에 따른 추가적인 이익까지도 기대할 수 있기 때문에 그 가능성이 서서히 인정되고 있는 추세이다.
이산화탄소의 전환기술은 크게 화학적 전환과 생물학적 전환으로 분류될 수 있는데 화학적 전환은 다음 Figure 1과 같이 분류할 수 있다. 그러나 그 기술적 특성에 따라 다시 열적 촉매화학적 전환, 광화학적 전환, 전기화학적 전환 등으로 나눌 수 있다.
또한, 이들 전환 공정을 기상 반응과 액상 반응으로 크게 구분할 수 있을 것이다. 기상반응은 열화학 또는 열촉매 반응 기술을 의미하며 액상 반응은 전기/광/바이오 화학전환 기술이
포함된다.
이산화탄소 전환 기술은 온실가스 저감 기술이 될 수 없다는 관점이 많다. 이는 전환 시 필요로 하는 많은 에너지로 인해 온실가스 발생을 유발한다는 측면과 전환을 통해서 얻어진 물질의 제품 주기가 짧아 다시 이산화탄소를 배출한다는 점이다. 따라서 이산화탄소를 자원화하는 CCU 기술의 도입, 특별히 이산화탄소의 화학적 전환 연구를 위해서 반드시 충족되어야 할 조건이 있는데, 첫째로는 이산화탄소를 재활용 하는 새로운 공정이 전체공정에서 총 이산화탄소의 발생량을 줄일 수 있어야 하고, 둘째로는 이산화탄소를 활용하는 새로운 공정이 기존 대체공정 대비 에너지 및 자원 사용량이 적어야 한다는 점이다. 현재 이산화탄소를 이용한 전환 기술 중 상용화가 이뤄진 공정은 포스겐과 같은 유해물질을 대체한 공정으로 폴리카보네이트 공정이 유일하다. 그러나 이산화탄소를 반응성을 갖는 일산화탄소, 카바메이트, 카보네이트 등으로 제조하면 많은 물질을 합성할 수 있다. 따라서 반응에 필요한 많은 에너지를 어디에서 얻느냐가 매우 중요한 문제이다. 그 해결방안으로 폐기물, 부생자원과 같은 미활용 자원/ 에너지 또는 태양에너지를 들 수 있다. 미활용 자원을 활용하는 경우 단순 열에너지를 얻는 경우에서 고부가 화학물질 원료로 활용이 가능하다. 전 세계가 일 년 동안 사용하는 에너지양은 지구에 들어오는 태양에너지의 한 시간 분량에 해 당한다고 한다. 이러한 태양에너지를 활용함으로써 추가적인 에너지의 투입 없이 이산화탄소의 화학적 전환을 가능하게 하는 광화학적 전환 반응이 친환경적인 미래 기술로 새로이 부각되고 있다. 따라서 본 총설에서는 이산화탄소의 화학적 전환 기술 중 투입되는 에너지의 양을 낮추어 상용화 가능성을 높이는 촉매화학적 전환과 외부에너지의 추가적인 투입을 필요로 하지 않는 태양광 에너지를 이용한 이산화탄소의 광화학적 전환에 관한 전반적인 연구 동향을 살펴보고자 한다. 본문 에서는 촉매화학적 전환 연구에 관하여서는 다양한 금속 촉매의 개발과 반응조건 및 공정 설계에, 그리고 광화학적 전환 연구에서는 빛에너지를 잘 흡수하거나 이산화탄소 전환효율을 높일 수 있는 광감응제 및 광촉매 개발에 중점을 두어 정리하였다.
[Table 1.] Use of carbon dioxide in the chemical industry for the synthesis of organic compounds[2]
Use of carbon dioxide in the chemical industry for the synthesis of organic compounds[2]
2. 열 촉매 화학적 전환 (thermochemical catalysis)
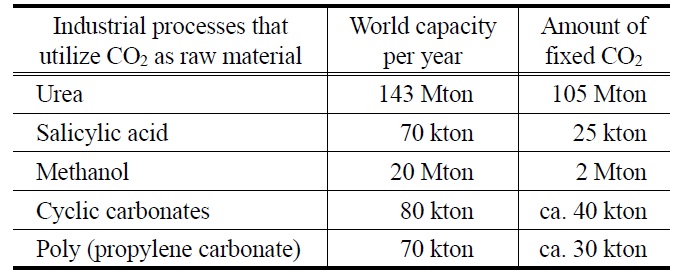
이산화탄소를 이용한 제품으로 가장 수요가 많은 제품은 Table 1에서 보는 것과 같다.
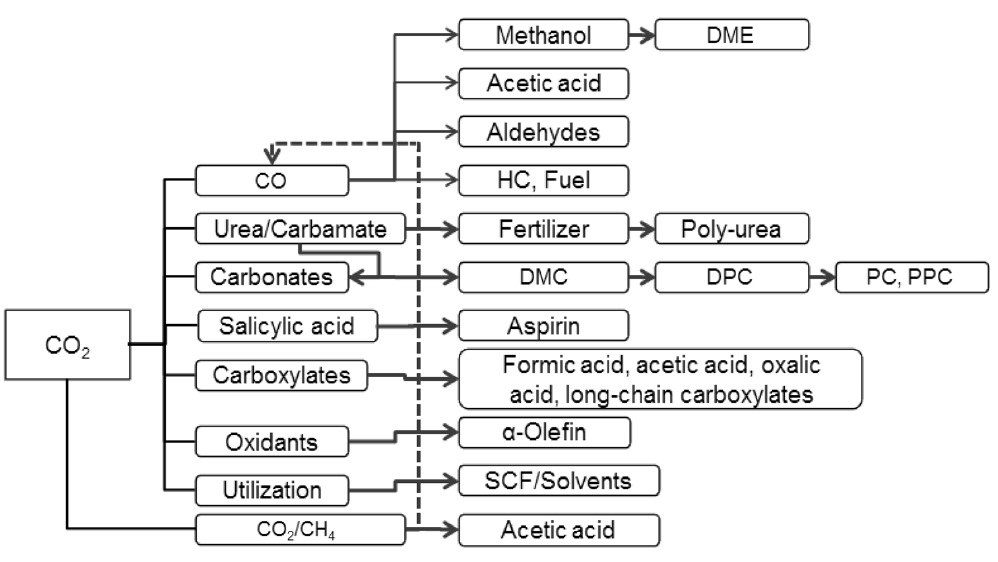
그외에 최근 여러 분야에서 널리 사용되고 있는 디메틸카보네이트를 들 수 있다. 현재 세계 디메틸카보네이트(dimethyl carbonate, DMC) 시장수요전망은 2010년 0.1~0.16 Gt/yr에 이르고 있으며 이들의 주 용도인 폴리카보네이트(polycarbonate, PC) 플라스틱 세계시장 규모는 1979년 0.15 Gt/yr에서, 1996년 1.0 Gt/yr, 2002년 2.6 Gt/yr로 급격히 증가되고 있다. 또한 내연기관 유류 첨가제인 MTBE (methyl tert-butyl ether) 대체제로서의 가능 시장규모는 1993년에는 0.06 Gt/yr에서, 2003년 30 Gt/yr으로 비약적인 성장이 예상되고 있으며 저독성 유기용 매, 정밀화학 중간체 시장, 이차전지의 전해액 세계시장에서 수십억톤에 이를 것이다. 매우 화학적으로 안정한 이산화탄소를 활용하기 위해서는 적절한 환원 물질이 필요하다. 그 예로 메탄을 비롯한 탄화수소, 수소, 암모니아를 비롯한 아민류, 이 중 결합 이상을 갖는 알킬류, 그리고 에폭사이드 등이 주요 환원제로 사용될 수 있으면 이를 통해 매우 다양한 플랫폼 화합 물을 만들 수 있고 이를 통해 다양한 고부가 제품을 만들 수 있다(Figure 2).
본 절에서는 단순 열분해 또는 촉매를 이용한 열분해 기술에 대해서 알아보고자 한다. 특히 건식개질, 이소시아네이트 및 알킬카보네이트 합성에 대하여 중점적으로 논하고자 한다.
본 반응은 메탄과 이산화탄소를 이용하여 일산화탄소와 수소를 합성하는 반응으로 주로 VIII족 전이 금속 촉매에서 일어 나는 것으로 알려져 있다. 본 반응이 주목 받고 있는 것은 이들 가스 모두 온실가스이며 매립지 가스(landfill gas, LFG) 또는 천연가스의 경우와 같이 함께 존재하는 경우가 많다는 것이 다. 물론 에탄, 프로판과 같은 다른 알칸화합물도 환원 원료로 사용이 가능하다.
2.1.1. 활성금속
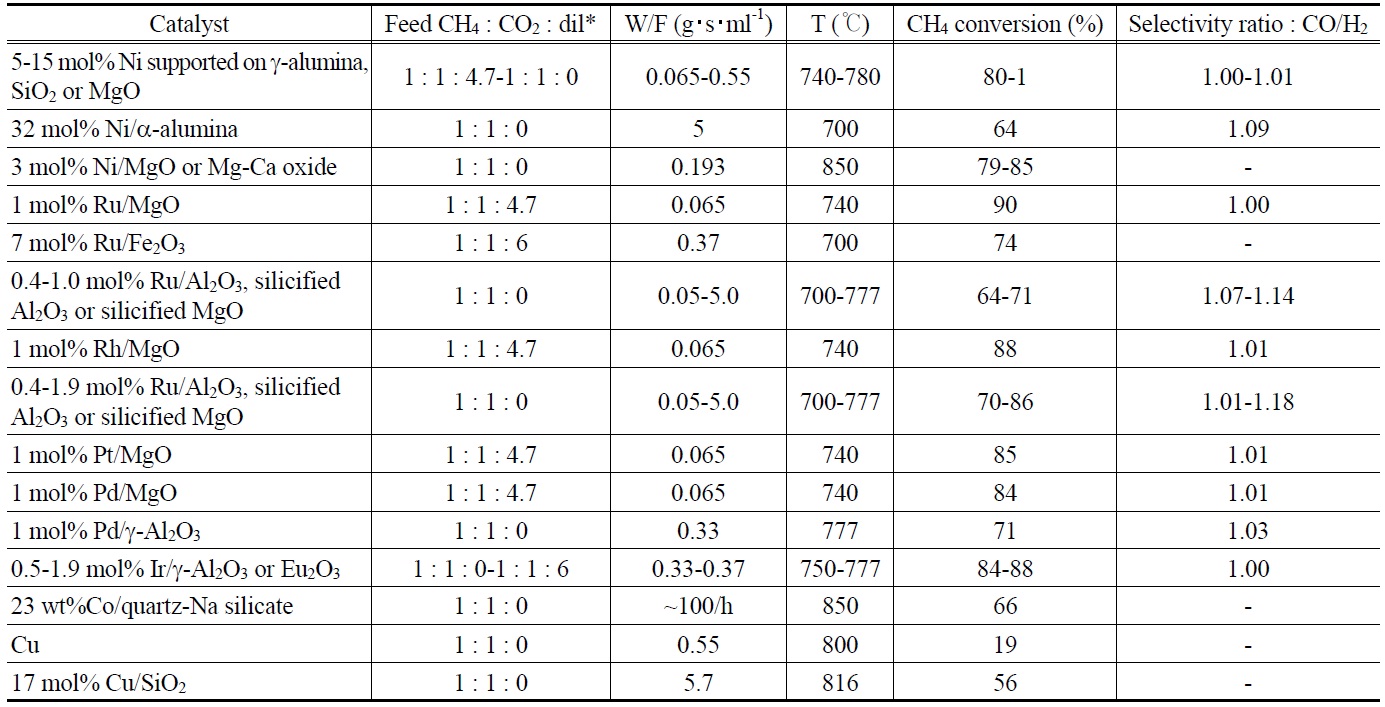
지금까지 연구결과를 토대로 이산화탄소/메탄 개질에 활성을 나타내는 촉매들의 성능을 Table 2에 나타냈다. Group VIII의 금속들의 환원형태가 담지되었을 때 활성을 가지는 것으로 보이며, 특히 그 중 Ni, Ru, Rh, Pd, Ir, Pr 등이 효과적인 촉매로 보고되고 있다.
미세한 MgO 단결정에 Ni, Ru, Rh, Pt, Pd을 담지시켜 평형 메탄 전환율이 각각 91, 90, 88, 85, 84%에 이르는 것으로 보고 하였다. 또한 CO/H2의 선택도 비는 약 1에 가까웠으며, 역수성 가스전이(reverse water gas shift, RWGS) 반응에 의한 수소의 손실도 크게 일어나지 않았다. 그밖에도 알루미나에 여러 금속을 담지시켰을 때 Rh > Pt & Pd > Ru > Ir 순서로 활성이 좋은 것으로 나타났다.
하지만 Pd와 Ru의 경우에는 낮은 활성과 낮은 수소 선택도를 보이는 것으로 나타났다. 또한 Ni과 Pd 촉매에서 탄소 생성이 일어났으며, 조사된 조건에서 Ni 촉매의 경우에서 보다 많은 코크가 생성되는 경향을 보였다. 대체적으로 Rh과 Ni 촉매가 활성이 좋고 장시간의 운전에 적합한 촉매로 알려져 있다. 그 중에서 고가인 Rh은 고 활성, 고 수소선택도, 무탄소
[Table 2.] Comparison of catalytic activity for carbon dioxide dry reforming of methane[3]
Comparison of catalytic activity for carbon dioxide dry reforming of methane[3]
운전 등 전체 성능에서 Ni보다 나을 수도 있다고 보고하였다. 또한 Rh 촉매는 폭넓은 공간속도에서 일정한 경향을 갖는 전환율 값을 보여주며, 이산화탄소와 메탄의 전환율 차이가 적고, 높은 수소 선택도에서 CO/H2의 비가 1에 가까운 결과를 보여주었다. 한편 Ni촉매는 활성은 좋지만 탄소생성의 심각한 문제가 있는 것으로 보고하고 있다.
지금까지 보고된 자료를 요약하면 Rh 담지촉매가 가장 활성이 좋은 촉매인 것으로 나타났으며, 상대적으로 경제적인 면에서 Ni 촉매가 주목을 받을 수 있지만 Ni 촉매의 사용을 위해서는 무탄소 운전을 위한 촉매의 개발과 운전 조건의 확립이 필요하다. Co, Ru, Pd, Ir, Pt 등의 Group VIII 원소들도 적절한 지지체에 담지된다면 효과적인 촉매역할을 할 수 있을 것으로 기대된다.
2.1.1.1. 코발트(Co)를 기반으로 하는 촉매
Ruckenstein and Wang[3]은 메탄의 건식 개질에서 산화마그네슘(MgO), 산화칼슘(CaO), 산화스트론튬(SrO), 산화바륨 (BaO), 감마 알루미나(γ-Al2O3) 그리고 실리카(SiO2)를 기반으 로 하는 Co 촉매의 활성도를 비교했다. 그리고 Co/MgO 촉매 에서 일산화탄소와 수소의 수율이 각각 93%와 90%에 도달 한다는 것을 보고했다. 이와 같이 높은 활성과 안정성은 CaO 와 MgO의 고용체에서 가능했는데, 이 고용체는 Mg와 Co의 상호작용을 강화시켰고 금속 소결과 탄소 침적을 예방했다. 그러나 γ -Al2O3에서 Co의 안정성은 Co의 양과 소성시키는 온도에 따라 변했다. 탄소 침적과 Co의 산화는 Co/γ -Al2O3의 비활성과 관련이 있는 것으로 알려졌다[4]. Hou and Yashima[5] 는 Co/α- Al2O3에서 Co의 표면과 개질 활성도 그리고 코크 형성 속도가 촉매의 소성 온도와 환원 조건에 매우 의존한다는 것을 보고했다. Bouarab et al.[6]은 MgO를 SiO2에 첨가하면 지지체의 작용이 메탄에서 수소를 추출하고 이산화탄소 흡착과 탄소 침적을 감소시킬 수 있기 때문에 Co/SiO2의 활성을 증가시킨다고 보고했다.
고순도의 CoOx-MgO는 이산화탄소를 이용한 메탄의 개질에 촉매화학적으로 비활성이라는 것 또한 알려졌다. 그러나 활성도와 상업적인 낮은 표면적을 가지고 있는 대공성의 실리카-알루미나를 기반으로 하는 CoOx-MgO의 수소 선택도(> 94%)는 명백하게 향상되었다[7]. 지지체의 전처리에 이용되는 금속 산화물의 촉매적 성능은 산화마그네슘(MgO) > 산화 지르코늄(ZrO2) > 산화세륨(CeO2) > 삼산화이트륨(Y2O3) > 이 산화티탄(TiO2)과 같은 순서를 따른다. TiO2를 기반으로 하는 Co 촉매가 850 ℃ 이하에서 환원될 때, 상당한 탄소 침적에 의해 비활성이 일어난다. 그러나 촉매는 처음 24시간 동안 자신의 활성도를 유지할 수 있고, 침적되는 코크의 양은 환원 온도가 850 ℃ 이상 올라갔을 때 상당해진다. Co/TiO2 촉매의 비활성화는 금속 코발트의 산화 때문이다. Co/TiO2에 첨가된 적은 양의 루테늄이 코발트의 산화를 억제하고 활성 사이트의 탄소 침적 또한 억제한다[8].
2.1.1.2. 귀금속 촉매
귀금속(Rh, Pt, Ir, Pd, Ru) 촉매는 탄소 침적에 대한 훌륭한 저항력 때문에 메탄의 건식 개질에 널리 사용되었다.
① 로듐(Rh) 촉매
Mark and Maier[9]는 메탄 전환은 금속 표면적이 증가해야 증가하고 담지체(γ-Al2O3, α- Al2O3, TiO2, ZrO2, SiO2, TiO2-SiO2, Al2O3-SiO2, ZrO2-SiO2)의 성격에 독립적이라고 주장했다. 반응 속도에서 기공의 구조 또는 담지체에 따른 효과는 발견할 수 없었다. 그러나 Wang and Ruckenstein[10]은 일련의 환원 가능한 산화물, 산화세륨(CeO2), 오산화니오븀(Nb2O5), 오산화 탄탈륨(Ta2O5), 이산화티탄(TiO2) 그리고 산화지르코늄(ZrO2) 과 환원 불가능한 산화물, 감마 알루니마(γ-Al2O3), 삼산화란 타늄(La2O3), MgO, SiO2, 그리고 삼산화이트륨(Y2O3)에서 Rh 의 촉매 성능을 비교했다. 로듐(Rh)의 담지체가 환원가능한 산화물일 때 일산화탄소와 수소의 수율이 낮다는 것이 발견 되기도 했다. Rh/MgO, Rh/γ-Al2O3에서 일산화탄소와 수소의 수율은 각각 83~85% 그리고 76~79%였다. Hou et al.[11]은 다 공성의 Al2O3를 기반으로 하는 Rh 촉매는 메탄의 건식 개질에서 높은 활성과 안정도를 가진다는 것 또한 발견했다.
② 백금(Pt) 촉매
Bradford and Vannice[12]는 메탄의 건식 개질에 사용되는 담체인 TiO2, ZrO2, Cr2O3, SiO2 중에서 TiO2와 ZrO2가 Pt 촉매에 좋은 지지체라는 것을 발견했다. 금속-지지체 상호작용과 Pt/TiO2 촉매의 유동적인 TiOx 종이 탄소 침적을 예방하기 때문이다. 1% Pt/ZrO2는 500시간 동안 심한 비활성이 없는 훌륭한 촉매이고, 촉매 활성은 Pt-ZrO2 주위에 있는 Pt의 접근성에 의해 결정되었다[13,14]. 그리고 ZrO2의 첨가는 Pt-Zrn 의 강한 상호작용 때문에 Pt/Al2O3의 안정성을 향상시키는데, 이 상호작용은 건식 개질에서 이산화탄소의 해리를 촉진시킨다[15]. Pt/Al2O3와 Pt/SiO2와 비교했을 때 SiO2 표면의 ZrO2와 Al2O3 표면의 La2O3는 Pt 촉매의 좋은 지지체다. 이 지지체는 염기 사이트에서 이산화탄소 흡착의 활성에 기여한다.
그러나 Bitter et al.[16]에 의해서 Pt 촉매의 비활성이 밝혀 졌다. 그들은 Pt 촉매의 비활성은 지지체와 금속의 입자 사이즈에 강하게 의존하고, 탄소 형성 속도는 Pt/γ-Al2O3 > Pt/TiO2 > Pt/ZrO2 순서로 감소한다고 했다. 큰 사이즈의 Pt 촉매(Pt 크기 > 1 nm)는 작은 사이즈의 Pt 입자를 가진 촉매에 비해 쉽게 비활성화 된다. 금속 소결보다는 탄소 침적이 비활성의 원인이라는 것이 밝혀졌다. Na 또는 K의 적은 양(특히 Na)은 염기 사이트를 증가시키기 때문에 Pt/Al2O3의 안정성을 증가 시킨다[17].
③ 이리듐(Ir) 촉매
Nakagawa et al.[18]은 건식 개질에서 이리듐 촉매의 담지체 의 활성은 다음과 같이 보고했다: TiO2 > ZrO2 >Y2O3 > La2O3 > MgO > Al2O3 > SiO2. 5%의 Ir/TiO2 촉매상에서 메탄과 이산화탄소의 전환율은 각각 27.6%, 37.9% 그리고 수소와 일산화탄소의 수율은 각각 22.4%, 32.7%였다. Ir/Ce0.9Gd0.1O2-x 촉매는 20시간 동안 상당히 안정했는데, 이것은 고체산화물연료전지 (solid oxide fuel cell, SOFC) 이용분야에서 바이오 가스의 직접 전환을 위한 양극 물질의 좋은 후보물질로서 제안되기도 했다[19].
④ 팔라듐(Pd) 촉매
5 nm의 평균 입자 사이즈를 가지고 있는 Pd/α-Al2O3 촉매 는 Pt/ZrO2 촉매와 비교할 만한 초기 활성을 보이고, CO/H2 ≒1과 수소의 높은 선택도를 보인다[20]. 그러나 24시간이 지나 면 촉매 활성은 50%나 감소하는데, 그 주요 원인은 탄소 형성과 금속 소결이다. Pd-CeOx/α-Al2O3에서 Ce의 첨가는 탄소 침적의 형성을 금지시키고 금속 소결을 감소시킨다.
⑤ 루테늄(Ru) 촉매
Ru/La2O3 촉매는 공급되는 이산화탄소와 메탄의 비율이 1 일 때, 550~650 ℃ 에서 80시간 동안 안정하다. 그러나 이산화탄소이 양이 증가하여 이산화탄소와 메탄의 비율이 1보다 크면, 과량의 이산화탄소 분위기에서 Ru의 부분 산화로 인해 Ru/ La2O3는 550 ℃ 에서 빠르게 비활성화 된다[21].
2.1.2. 촉매 지지체
Group VIII 금속들은 지지체에 담지되어 사용되며, 백금족의 경우는 알루미나, 마그네시아, 유로피아 등의 지지체에 담지되었을 때 높은 활성을 보인다. 니켈(Ni) 촉매의 경우에는 이밖에 실리카, 티타니아 혹은 실리카-알루미나, 마그네시아- 알루미나, 칼시아-알루미나와 같은 혼합 산화물 등에 담지되었을 때 좋은 결과를 나타냈다.
Ni/γ -Al2O3 촉매의 사용시 야기되었던 탄소의 생성은 알루미나를 마그네시아로 대체하여 촉매의 안정성을 향상시킴으로써 줄어드는 경향을 보였다. 이러한 결과는 지지체가 탄소 생성점인 금속 클러스터의 형태를 제어함으로써 탄소의 생성에 영향을 미치는 것으로 판단된다.
일반적으로 촉매의 담지량이 높을 때는 금속-지지체 상호작용이 낮은 담지량에서 만큼 주요하지 않다. 예를 들면 Rh/SiO2 촉매에 Al2O3, TiO2, MgO 등을 첨가하였을 때 CO2/CH4 개질 반응과 RWGS 반응이 촉진되는 반면, 메탄의 부분산화 반응 이나 CO2/CH4 반응이 촉진되지 않는 것으로 보고하고 있다. 따라서 이러한 CO2/CH4 반응이 촉진되는 현상은 금속의 분산도나 금속-지지체 상호작용에 의한 것이라기보다는 이산화탄소의 활성화와 이산화탄소의 흡착력을 강화시키는 염기성 산화물의 역할 때문으로 해석된다. 즉, 염기성 산화물에 카보네이트나 포매이트 형태로 흡착된 이산화탄소가 활성점이 Rh으로 이동하여 해리된다. 이러한 해석은 알칼리금속이 함유된 백금족 금속들의 이산화탄소 활성화 메커니즘과 유사하다. 또한 몇몇 산화물 지지체는 촉매 표면에 형성된 탄소의 반응성에 영향을 미치는 것으로 알려졌다. 서로 다른 지지체에 담지된 Rh 촉매에 생성된 탄소를 온도 상승에 따라 수소 첨가반응을 행하면 SiO2를 지지체로 사용한 경우보다 Al2O3와 TiO2를 지지체로 사용하였을 때 탄소의 반응성이 좋은 것으로 보고하고 있다.
2.1.3. 건식개질의 반응메커니즘
니켈 촉매의 반응메커니즘을 예로 들면 다음과 같다. 메탄의 건식 개질 반응메커니즘은 Bodrov and Apel'baum[22]이 처음에 제안했다. 이산화탄소는 RWGS 반응에 의해 일산화탄소로 전환되고(CO2 H2 → CO H2O), 수소와 일산화탄소 또한 H2O와 CHx의 반응을 통해 생성된다. 이 메커니즘은 연속적으로 진행되고 몇몇의 가능한 다른 반응메커니즘이 보고되고 있다.
니켈을 기반으로 하는 촉매에서, 개질 반응은 활성사이트의 표면에서 메탄의 분리 흡착을 통해 일어난다는 것이 널리 인정되기도 하였다(*는 니켈표면을 의미함).
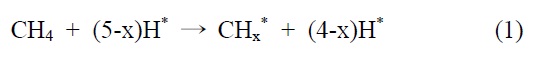
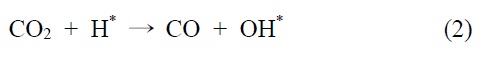
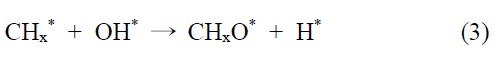
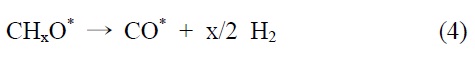
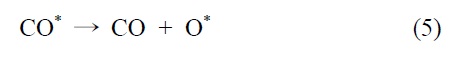

이산화탄소는 니켈 표면에서는 해리되지 않는 것이 확인되었기 때문에 메탄 분해에서 파생된 수소는 (식 (1)) Ni/SiO2 에서 이산화탄소의 흡착과 활성화를 위해 필수적이다. 또한 Ni/SiO2 촉매에서, 속도 결정 단계가 어떤 반응인지가 주요 논쟁거리이기도 하다[23]. Osaki et al.[24]은 Ni/SiO2, Ni/MgO, Ni/Al2O3 그리고 Ni/TiO2에서 메탄 건식 개질의 메커니즘을 조사하기 위해 표면 반응 분석을 이용했다. 이 결과에 의하면 수소를 생산하고 메탄의 흡착과 분해가 CHx 종을 형성한다 (식 (1)). 그 다음 CHx와 이산화탄소 또는 산소(식 (3))의 표면 반응이 속도 결정단계라고 주장했다.
반면에, Hu and Ruckenstein[25]은 NiO/SiO2 촉매상에서 비활성을 나타내는 C와 O 사이에서 일어나는 반응메커니즘은 다음과 같다고 보고하였다.
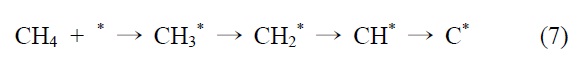
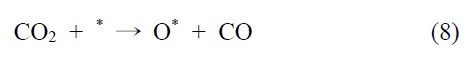
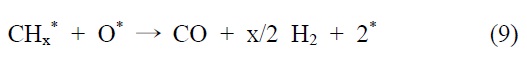
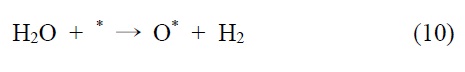
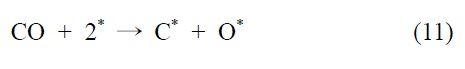
2.2. 아민과 이산화탄소를 이용한 이소시아네이트 합성
이소시아네이트(R-NCO)는 플라스틱, 수지, 각종 첨가제 등의 제조에 가장 많이 사용되는 원료 중의 하나이다. 예를 들면 디이이소시아네이트(diisocyanate)는 폴리우레탄의 원료로 오래전부터 사용되어오고 있고, 지난 2000년의 경우만 보더라도 4백만톤 규모의 시장을 형성하며 성장하였다. 수요를 분석해 보면 메틸렌 디페닐 디이소시아네이트(methylene diphenyl diisocyanate, MDI)가 61.3%로 가장 많이 사용되고 있고, 톨루엔 디이소시아네이트(toluene diisocyanate, TDI) 34.1%, 헥사메틸렌 디이소시아네이트(hexamethylene diisocyanate, HDI) 3.4% 등으로 순으로 되어 있다[26].
일반적으로 이소시아네이트를 제조하는 방법은 다양하다. 우레탄의 열분해, N-알킬카바마이트(N-alkylcarbamate)와 삼 염화붕소/염화보란/염화실란(BCl3/chloroborane/chlorosilane) 의 반응이 있다. 하지만 상업용으로 제조되는 경우에는 아직까 지 아민 포스겐반응(amine phosgenation)이 유일하다[27-29]. 그렇지만 항상 포스겐(phosgene)의 위험성과 안전성에 대해서 논쟁의 대상이 되고 있어 이를 대체하기 위한 방안이 논의 되고 있다.
한 가지 방안으로서 이산화탄소를 원료로 하는 이소시아네이트 합성반응이 있다. 이것은 헤테로큐물렌(heterocumulene) 의 탈산소반응에 의해 진행되며 다양한 금속계 균일촉매가 배위공간을 변형시키면서 반응을 촉진시킨다. 1976년 Tsuda et al.[30]에 의해 구리 t-부톡사이드(copper(II)
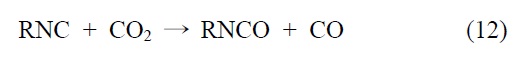
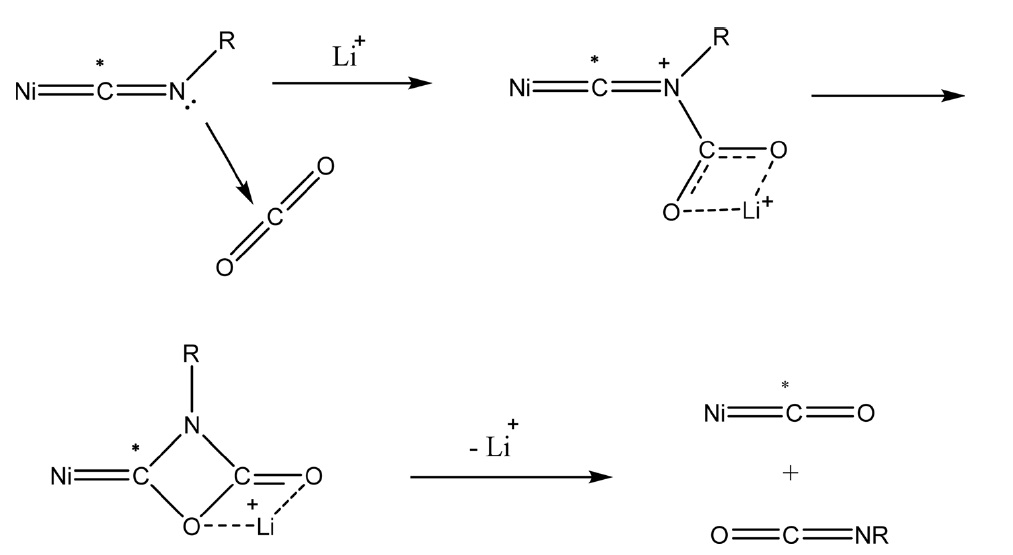
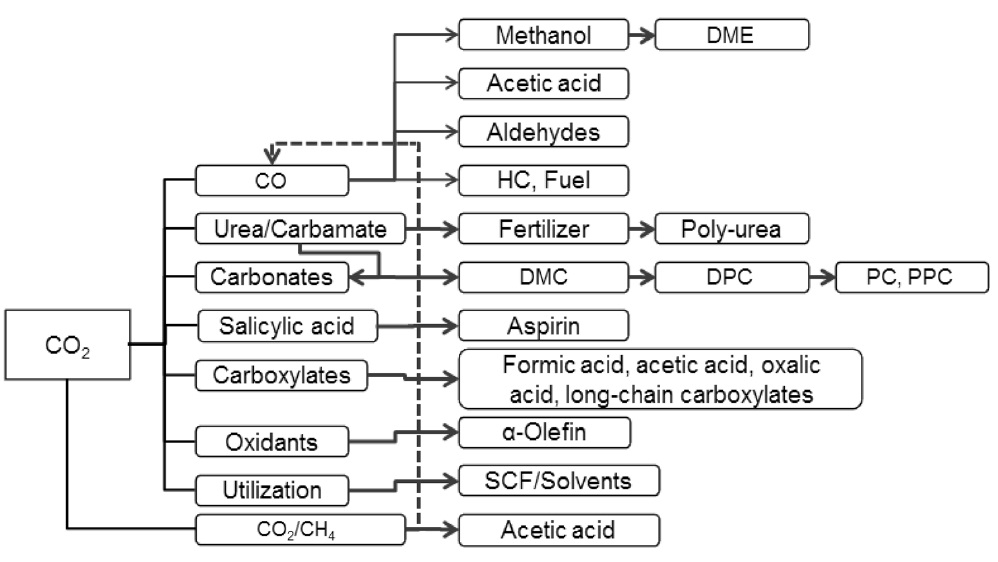
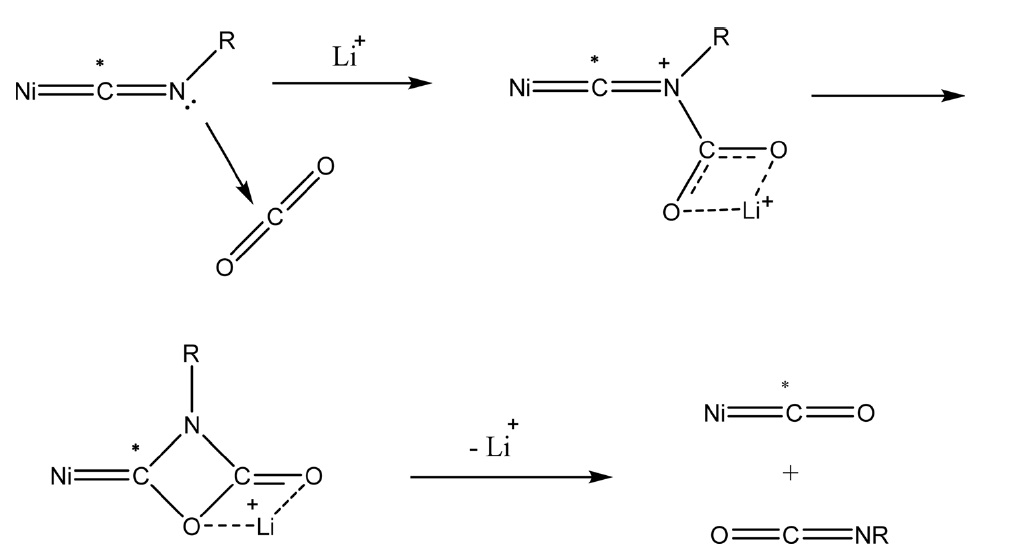
유사한 반응으로 Ni(NCR)4 (R = Me, 2,6-Me2C6H3) 상에서 이산화탄소의 반응에 의해 Ni(CO)2(CNR)2와 RNCO를 제조하는 방법이 보고되었다. 이 경우는 알카리 금속이온, 특히 Li 이온에 의해 반응이 활성화되어 Figure 3과 같이 다중결합 복분해(multiple bond metathesis) 반응이 일어나는 것으로 알려져 있다[31].
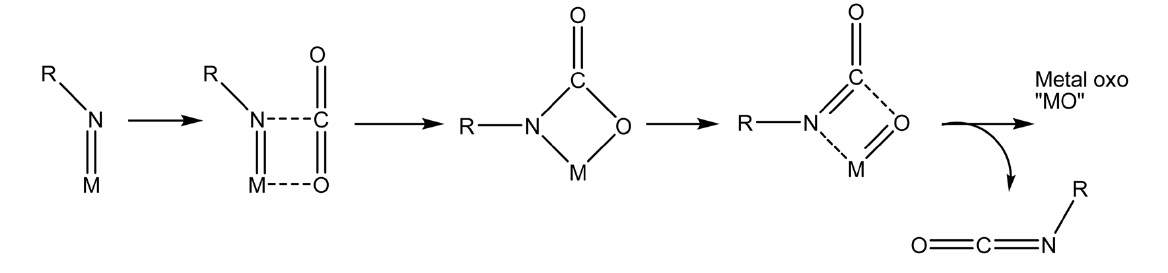
이산화탄소와 금속-아릴아미도 복합체(metal(Ti(V), U(V)) arlyamido-complex)의 반응에 의해서도 아릴이소시아네이트 (arylisocyanate)를 제조할 수 있다. 이 반응은 상압, 상온에서 진행된다는 특징이 있으며 금속이 불필요한 산소를 잡아주는 역할을 하는 [22] 첨가환화(cycloaddition), 금속-이미도 복합체(metal-imido complex)생성 등의 반응에 의해 진행되는 것으로 보고되고 있다[32](Figure 4).
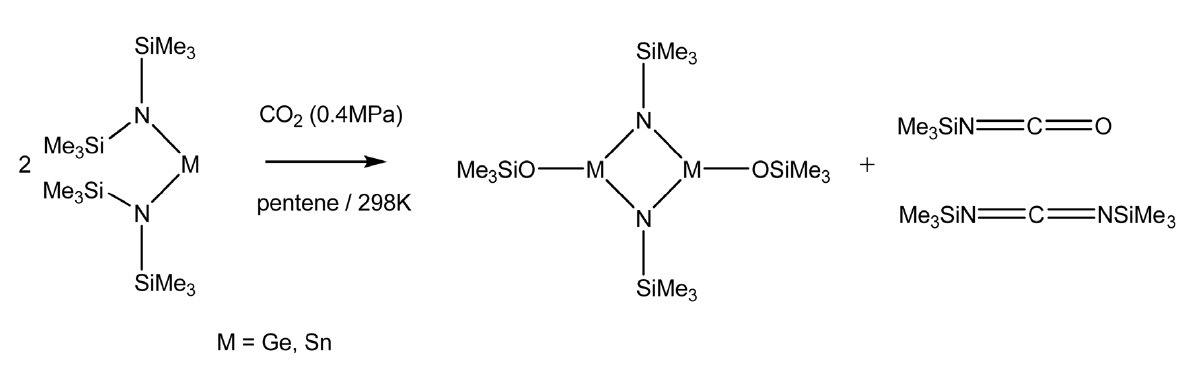
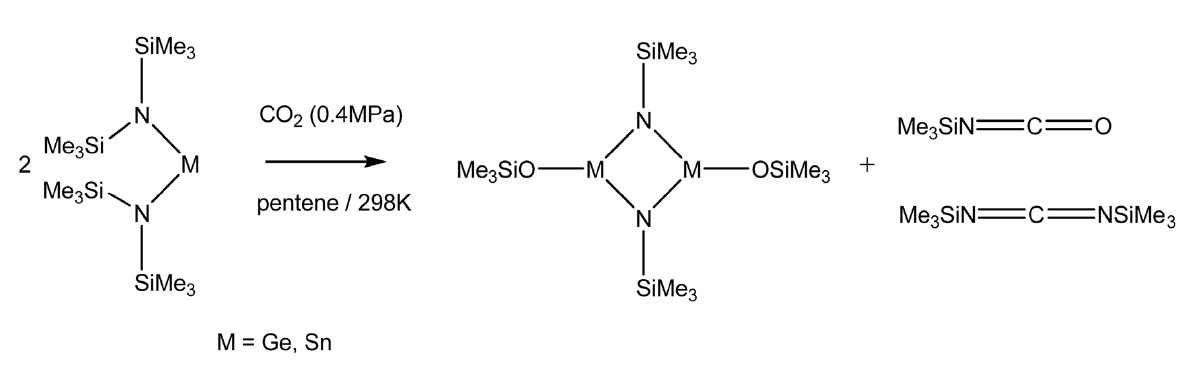
또한 실릴 이소시아네이트(silyl isocyanate)를 제조하기 위해 이산화탄소와 리간드의 실릴기(silyl group)의 옥소 이동(oxotransfer) 반응을 이용한 경우도 있다. Sita et al.[33]은 이 반응에 group XIV의 Ge, Sn 비스아미드(bisamide)를 사용할 경우 수율과 선택도를 크게 높일 수 있다고 보고하고 있다(Figure 5).
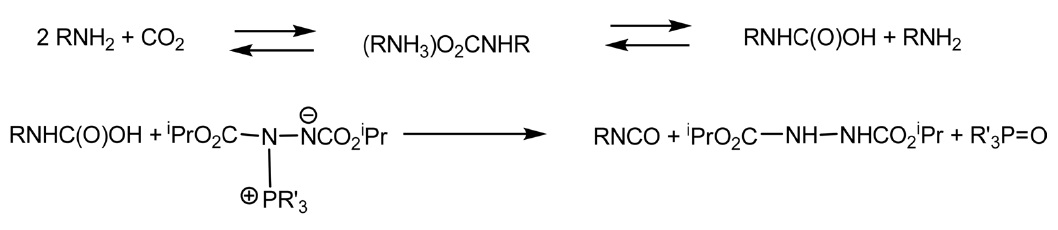
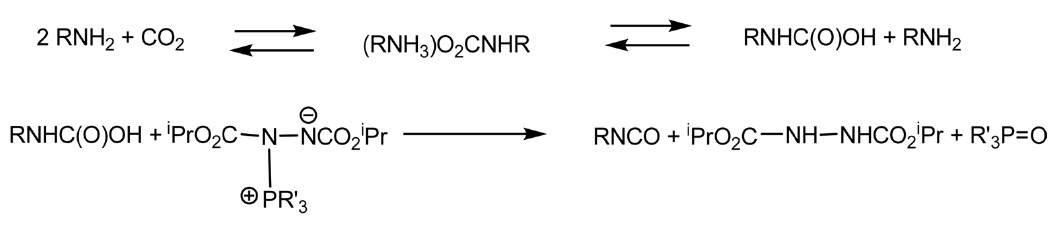
그밖에도 이산화탄소와 아민에 의한 이소시아네이트 직접 합성방법이 소위 “미즈노부 화학(Mitsunobu chemistry)”을 이 용하여 제안되기도 했다. 원래 이 반응은 정반응 보다는 역반응으로 평형이 이동하지만, 정반응으로 진행되는 조건을 찾 는다면 높은 수율을 얻을 수 있다는 관점에서 시작되었다. 즉 미즈노부 양쪽성이온(Mitsunobu zwitterion)을 이용하는 것으로 아래 반응식과 같이 진행되며 일차 알킬아민의 경우 높은 수율을 나타내지만 방향족 아민에서는 수율이 낮다는 단점이 있다. 하지만 POCl3, PCl3, P4O10, SO3, SOCl2와 같은 탈수제 (dehydrating agent) 등을 사용한다면 수율 증가에 도움된다는 보고가 있다[34,35](Figure 6).
이상과 같이 여러 가지의 이소시아네이트 제조방법이 보고 되어 있지만 상업적으로 활용하기 위해서는 경제성 부분과 함 께 환경적인 요소 등에 대해 충분히 극복할 수 있는 방안이 제시돼야 한다. 상기에서 보듯이 현재 이산화탄소를 이소시아 네이트 합성원료로 활용하고자 하는 기술이 기초단계에 와 있으나 많은 연구자들의 관심과 노력에 의해 더욱 발전되리라 전망된다.
이산화탄소를 원료로 하여 알킬카보네이트를 제조하는 기술은 크게 두 가지, 간접 전환과 직접 전환으로 나눌 수 있다.
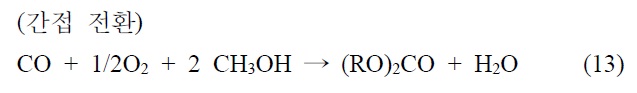
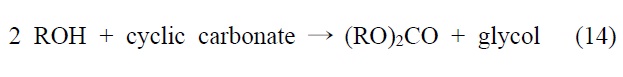
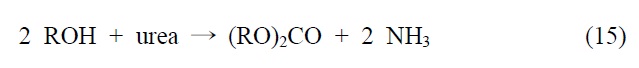
이 반응은 반응조건에 따라 카보네이트의 수율과 선택도를 증가시킬 수 있어서 상업적으로 유용하게 활용하고 있다.
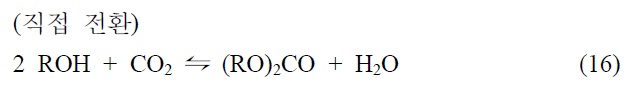
직접 전환반응은 열역학적으로는 평형상태에 있어서 간접 전환에 비하여 수율이 낮고 반응조건이 심한 편이다. 하지만 실제 상용화하기 위해서 부생성물에 대한 개념이 중요하다. Table 3에서 보는 바와 같이 여섯 가지의 카보네이트 제조방법을 비교했을 때 HCl, glycol, NH3, H2O 등이 생성된다. 녹색화학적인 측면에서 판단하기 위해 “그린화학 원칙(green chemistry principles)” 개념에서의 기준인 AE 지수(atomic economy factor: a measure of the efficiency of a reaction, wt%로 표시) 를 도입한다. 즉 다섯 가지의 화학양론적인 반응에서 제조한 디메틸카보네이트의 AE값은 반응될 때 사용되는 원자(atom) 와 사용되지 않는 원자를 구분하여 백분율로 나타내는 것이다. Table 3에서 비교했을 때 phosgene에 의한 제조방법은 AE 값이 55 wt%로서 디메틸카보네이트 1톤 생산할 때 0.8톤의 HCl이 생성된다는 것으로 의미한다[36,37].
에틸렌카보네이트의 메타놀분해(methanolysis)에 의한 경우 (2번) AE값은 59%로서 글리콜(glycol)이 생성되는 반응이
[Table 3.] AE factor for various DMC routes[36]
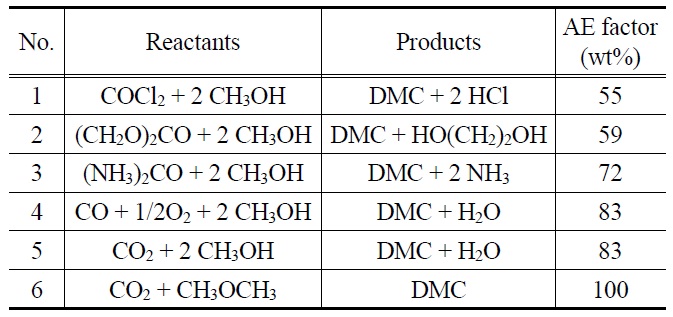
AE factor for various DMC routes[36]
다. 카보네이트 제조에 있어서 이산화탄소를 활용하는 기술은 “비 포스겐 기술(non-phosgene technology)”로서 많은 관심을 갖는 분야이기도 하다.
2.4.1. 에틸벤젠 탈수소화 반응
이산화탄소를 산화제로 활용하는 경우로 대표적인 공정이 에틸벤젠의 탈수소화 반응을 들 수 있다. 에틸벤젠 탈수소화를 통하여 스티렌을 얻을 수 있다. 스티렌의 일반적인 제조 공정은 벤젠을 에틸렌을 이용하여 알킬화 반응을 이용하여 제조 되고 있으나 본 기술이 개발되면서 이산화탄소 활용 기술로 주목받고 있다. 본 반응은 다음 반응과 같이 진행된다.
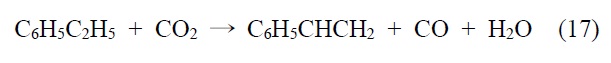
이산화탄소에 의한 탈수소화는 해외에서도 90년대 들어 연구 결과가 발표되기 시작하여 일본 자원환경 연구소, 미쯔비시 케미칼 등에서 촉매 및 상업화 연구가 진행 중에 있으며 기타 선진국에서도 이들 연구가 진행 중에 있다. 국내의 경우에도 한국화학연구원(KRICT), 한국과학기술원(KAIST), 포항공대 (POSTECH), 한국과학기술연구원(KIST), 한국에너지기술연구 원(KIER) 등 여러 기관에서 연구되어 왔다. KIER에서는 파일롯 플랜트(Pilot Plant) 연구(SM 250 kg/day)가 이뤄졌으며 KRICT/삼성종합화학에서 역시 파일롯 플랜트 연구(SM 100 kg/day)가 수행되고 있다.
2.4.2. 탄화수소 합성 반응
본 기술은 합성 원유 제조 기술로 석탄 청정 연료화 연구와 매우 밀접한 기술이다. 이산화탄소를 탄화수소로 전환하는 연구로 고유가 시대에 연구되어 온 분야이다. 이산화탄소의 탄화수소 전환 기술은 일산환탄소를 제조하여 이를 피셔-트롭쉬 합성반응(Fischer-Tropsch synthesis, FTS)을 거쳐서 합성원유 를 제조하는 방법과 메탄올을 합성하여 합성원유를 제조하는 기술로 나뉘어진다.
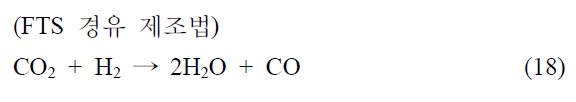
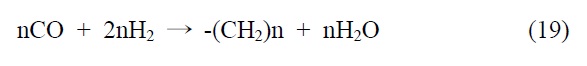
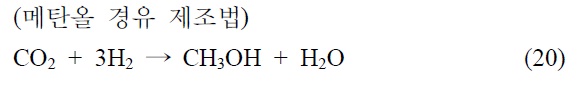
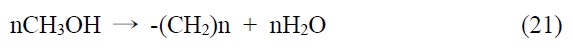
본 기술의 해외 동향을 알아보면 일본 오사카 국립연구소 및 교토대학 등에서 메탄올 경유법으로 제조하고 있으나, C2~4 가 주로 생성되므로 부가가치가 낮은 것으로 알려져 있다. 독 일 Karlsruhe 대학 FTS 경유법으로 Fe/Cu/Al2O3 촉매사용 공정개발되었으며 미국 Exxon에서도 FTS법에 의한 촉매 공정이 개발된 것으로 알려져 있다. 국내의 경우 KAIST FTS 경유법 촉매 반응공정에 필요한 촉매 기초연구가 이뤄졌으며 현대오 일뱅크에서는 실험실적으로 80% 탄화수소 선택성 촉매공정개발(FTS)이 개발된 것으로 알려져 있다.
3. 광화학적 전환(photochemical catalysis)
인공광합성(artificial photosynthesis)으로 대표되는 광화학적 이산화탄소 전환 연구는 자연계에서 태양광을 에너지원으로 사용하여 물과 이산화탄소로부터 당류로 대표되는 고에너지 유기물을 합성하는 광합성 반응에 그 기초를 두고 있다. 광합 성 반응은 식 (22)에서 보는 바와 같이 전체적으로 이산화탄소와 물에서 태양광을 이용하여 유기물 및 산소를 발생시키는 반응인데, 자연계에서는 녹색 식물이나 녹색 박테리아 내에 서 이러한 광합성이 활발히 일어남을 볼 수 있다. 광합성 반응은 크게 명반응과 암반응 두개의 과정으로 구성되어 있는데 명반응에서는 식물이 엽록체를 이용하여 빛을 흡수한 뒤 물을 분해하여 산소와 수소를 생성하고, 암반응에서는 얻어진 수소를 이용하여 이산화탄소를 환원하여 최종적으로 유기물을 합성하게 된다. 이러한 자연계의 광합성 반응을 모방하여 태양광을 에너지원으로 사용하여 이산화탄소와 물을 산소와 액체연료로 전환하려는 연구가 바로 인공광합성 기술이다. 따라서 인공광합성 기술은 빛에너지를 이용하여 물을 분해하여 수소와 산소를 생성하는 연구와 이산화탄소를 유용한 화합물로 전환하는 두 가지 연구를 모두 포함하고 있으나 본 총설에서는 빛에너지를 이용한 이산화탄소의 직접적 광환원 기술 부분만을 살펴보고자 한다.
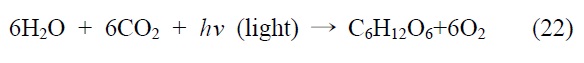
태양광에서 에너지를 수집하여 화학에너지로 전환하는 자 연계의 시스템은 매우 복잡하고 다양하나 기본적으로는 빛을 수집하는 광수집 안테나 복합체(light harvesting antenna)와 광합성 반응 중심(photosynthesis reaction center)으로 이루어져 있다. 빛을 수집하는 안테나의 경우 많은 색소분자로 구성되어 있으며 빛에너지를 흡수한 후 반응 중심으로 여기된 에너지를 전달하는 역할을 담당한다. 인공광합성 기술로 표현되는 이산화탄소의 광화학적 전환 시스템 역시 이와 유사하게 빛에너지를 흡수하는 광감응제(photosensitizer) 부분과 광감응제에서 흡수된 빛에 의하여 여기된 전자가 이동하여 이산화탄소를 환원시키며 다양한 부산물을 생성해내는 반응중심에 해당하는 촉매(catalyst) 부분으로 구성되어 있으며 경우에 따라서 하나의 물질이 광감응제와 촉매의 두 가지 역할을 모두 담당하기도 한다. 따라서 태양광의 에너지를 효과적으로 수집할 수 있는 안정적인 광감응제의 개발이 이산화탄소의 광화학적 전환 연구의 주된 관심사 중 하나라고 볼 수 있겠다.
아울러 광화학적 이산화탄소 전환 반응은 빛에너지에 의해 여기된 전자를 이용하여 이산화탄소를 환원시키는 반응으로, 반응에 사용되는 전자(electron)와 양성자(proton)의 개수에 따라 다양한 환원반응과 생성물이 존재하게 되는데, 그에 따른 이산화탄소 환원 전위와 생성물을 살펴보면 다음과 같다(pH 7, 수용액상에서 표준수소전극 기준).

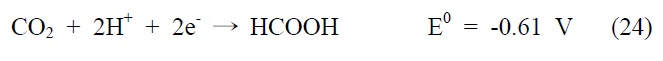
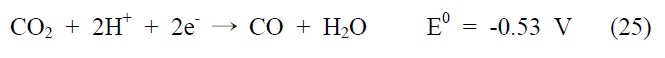
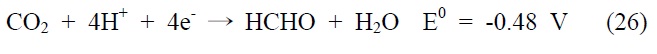
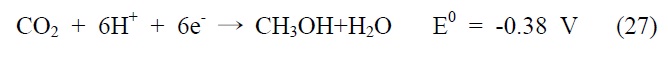
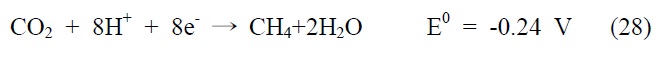
식 (23) 반응은 이산화탄소와 하나의 전자가 반응하는 단전 자 전달(one-electron transfer) 반응으로, 환원 전위값이 매우 높아 반응이 진행되기 무척 어렵고 생성물 또한 불안정한 이산화탄소의 음이온 라디칼로 존재하게 된다. 이에 상대적으로 환원 전위값이 낮고, 안정하면서 메탄올과 같이 부가가치를 가진 부산물을 생성하는 식 (24)~(28) 반응들이 실제적으로 더 중요한 반응으로 여겨지고 있는데, 이 반응들은 모두 여러 개의 전자와 양성자가 한꺼번에 반응에 참여해야 하는 다전자 전달(multi-electron transfer) 반응이다. 따라서 이산화탄소를 표면에 안정적으로 결합시켜 전달된 전자를 이용한 다전자 전달 반응, 그 가운데서도 메탄올 합성과 같은 특정 다전자 전달 반응을 선택적으로 가능케 하는 촉매의 개발 역시 광화학적 이산화탄소 전환 연구의 주된 관심사라고 할 수 있겠다.
이와 같이 광화학적 이산화탄소 전환기술 연구는 사용되는 광감응제와 촉매의 분류적 특성에 따라 크게 1) 주로 전이 금속 착물(transition metal complex)을 이용한 균일계 반응 시스템(homogeneous reaction system)과 2) 무기물질인 반도체 광 촉매(inorganic semiconductor photocatalysts)를 이용한 비균일계 반응 시스템(heterogeneous reaction system), 그리고 3) 균일계와 비균일계 등 다양한 광촉매가 접목된 혼성 시스템(hybrid) 시스템으로 나눌 수 있으며, 여기에 추가적으로 4) 광촉매 물질을 전극으로 활용하여 광에너지와 전기에너지를 동시에 가해주는 광전기화학 반응 시스템(photoelectrochemical reaction system) 관련 연구도 포함시킬 수 있다. 이에 본 단락에 서는 위에서 언급한 4가지 반응 시스템으로 나누어 관련 연구 결과들을 정리하였다.
3.1. 균일계 반응 시스템(homogeneous reaction system)
태양광을 에너지원으로 사용하여 이산화탄소를 광화학적 으로 환원시키는 반응 가운데 균일계 반응은 주로 전이금속 화합물로 이루어진 분자 촉매를 중심으로 연구가 진행되고 있다. 앞서 설명했던 바와 같이 이산화탄소의 광화학적 전환 시스템은 크게 빛에너지를 흡수하는 광감응제(photosensitizer) 부분과 광감응제에서 흡수된 빛에 의하여 여기된 전자가 이동하여 이산화탄소를 환원시키는 촉매(catalyst) 부분으로 나 눌 수 있는데, 균일계 반응의 경우 주로 전이금속 화합물로 이루어진 분자 광감응제와 분자 촉매가 결합된 이성분계 연구가 대부분이나 특정 분자의 경우 두가지 역할을 모두 담당하여 단일성분계가 구성되기도 한다.
3.1.1. 단일성분계(single component system)
단일성분계 분자 촉매는 한 분자가 광감응제(photosensitizer, P)기능과 촉매(catalyst, Cat)기능을 모두 담당하는 시스템으로, 단일성분계에서의 이산화탄소 전환반응은 식 (29), (30)에 서와 같이 분자촉매와 용액상에 첨가된 전자공여체(electron donor)의 결합이 먼저 이루어진 후, 조사된 빛에너지에 의해 분자촉매와 전자공여체의 결합체가 여기됨과 동시에 결합이 분리되면서 환원된 형태의 분자촉매와 산화된 형태의 전자공 여체로 나누어지며, 이 때 생성된 환원된 형태의 분자 촉매에 의해 이산화탄소가 환원되는 과정을 통해 일어나는 것으로 알려져 있다. 대표적인 단일성분계 분자촉매의 구조는 Figure 7에 표시하였다.
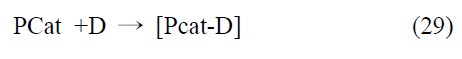
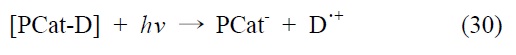
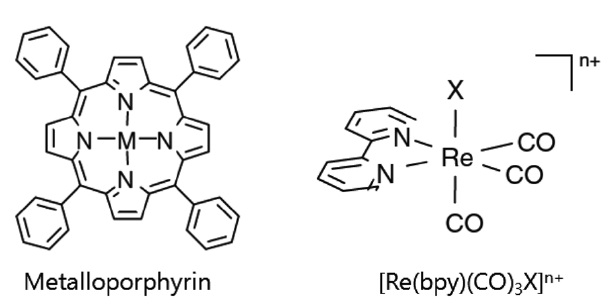
3.1.1.1. 금속 포피린 및 거대분자 화합물
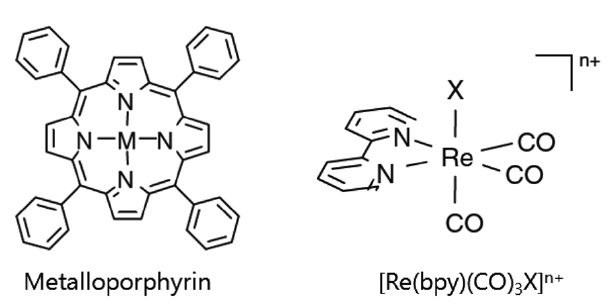
금속 거대분자 화합물(metallomacrocyclic compounds)은 가시광 영역에서 매우 큰 흡광계수를 가지고 있어 효율적인 광에너지의 포획이 가능하며 동시에 거대분자 화합물과 결합된 중심 금속이 환원되면서 활성부위(catalytic active site)로 작용하여 이산화탄소의 환원을 가능케 하는 장점을 가지고 있다. 금속 포피린(metalloporphyrin)을 포함한 금속 거대분자 화합물에 관련한 연구는 주로 Grodkowski et al.에 의해 이루어졌는데, 이때 사용된 금속 거대분자 화합물은 metalloporphyrin, metallocorrins, metallophthalocyanines, metallocorroles 등으로 철 (Fe)과 코발트(Co)가 중심금속으로 사용되었다[38-42]. 금속 포피린에서의 이산화탄소의 광화학적 환원 반응은 1) 전자공 여체(electron donor)로 첨가한 트리에틸아민(triethylamine, TEA)
이 포피린에 리간드 형태로 결합한 뒤 2) 광조사하에 빛에 의 해 여기된 TEA˙+가 떨어져 나가는 과정을 통하여 3) 3가 형태로 존재하던 금속포피린(MIIIP)이 0가의 금속포피린([M0P]2-) 로 전환되고, 4) [M0P]2- 형태의 금속포피린이 포피린과 결합 되어 있는 이산화탄소를 환원시키며 최종적으로 일산화탄소 (carbon monoxide) 혹은 개미산(formic acid)을 부산물로 생성하는 것으로 밝혀졌다. 그러나, 금속 거대분자 화합물의 경우 빛을 흡수한 뒤 여기된 전자의 라이프타임이 매우 짧기 때문에 촉매로서의 활성부위가 매우 적게 생성되므로 결과적으로 광효율 및 촉매적 활성이 낮은 단점이 있다.
3.1.1.2. Re 카르보닐 화합물(Re(CO)3(bpy)X Complexes)
Re 카르보닐 화합물을 이용한 광화학적 이산화탄소 환원은 Hawecker et al.[43]에 의해서 처음으로 보고되었다. 이들은 ReIL(CO)3X (L = 2,2'-bipyridine or 1,10-phenanthroline, X = Clor Br-) 화합물과 트리에탄올아민(triethnolamine) 전자공여체 를 사용하여 광조사하에 이산화탄소가 일산화탄소로 선택적 전환되는 것을 확인하였다. 이후 Ishtani et al.[44-47]을 중심으로 다양한 리간드로 치환된 Re 카르보닐 화합물이 합성되어 이산화탄소의 광화학적 환원 반응에 적용되었는데, 치환된 리간드에 따라 Re 화합물의 전기적특성 및 이산화탄소 환원 반응에 대한 활성도 크게 달라지는 것으로 나타났다. 예를 들 면 [Re(bpy)(CO)3{P(OEt)3}]+ 의 경우 두배 가까이 증가된 이산화탄소 전환효율을 보였으나, [Re(bpy)(CO)3(py)]+ 혹은 [Re(bpy) (CO)3(PPh3)]+ 화합물의 경우는 오히려 그 활성이 감소되는 것으로 나타났다[44,45]. 또한 피리딘 리간드에 CF3와 같은 전자를 끄는 그룹이 존재할 경우 Re 화합물의 활성이 현저히 떨어지는 반면 MeO와 같은 전자를 주는 그룹의 경우는 활성에 별다른 영향을 미치지 않는 것도 확인되었다[46,47]. Re 비스카르보닐 화합물을 사용했을 경우에도 비슷한 양상을 보였는데, 일례로 [Re(dmb)(CO)2(PR3)(PR’3)]+ (dmb = 4,4’-dimethyl- 2,2’-bipyridine) 화합물의 포스핀 리간드에 전자를 주는 그룹인 FPh (fluophenyl)기가 함유되어 있을 때 매우 효과적으로 이산화탄소가 일산화탄소로 전환되었다[48,49]. Re 화합물을 이용한 이산화탄소 환원반응에서는 부산물로 일산화탄소가 선택적으로 생성됨과 동시에 수소는 거의 생성되지 않는 것으로 나타나 이산화탄소 환원반응의 선택성이 다른 광촉매 물질에 비하여 매우 우수한 것으로 알려져 있다. 또한 Re 화합물의 광효율은 단일성분계의 경우는 최고 38%, 그리고 각기 다른 Re 화합물을 함께 사용한 이성분계의 경우 최고 59%로 다른 균일계 촉매에 비해 광분해효율이 매우 높은 편이다[44,46]. 그러나 Re 화합물의 광분해 반응도 동시에 진행될 수 있기 때문에 촉매 활성은 낮다고 볼 수 있다.
Re 화합물에 의한 이산화탄소 환원 반응은 광조사에 의한 금속-리간드 전하이동(metal to ligand charge transfer, MLCT)에 의해 유발되는 것으로 알려져 있으며, Re 화합물의 할로겐 리간드(X)가 용매 분자로 치환되어 Re 화합물과 용매분자가 결합 된 형태의 중간체 혹은 용액 상에 존재하는 양성자가 치환된 형 태의 Re-hydride 중간체 등이 형성된 후 중간체와 이산화탄소와의 결합을 통해 이산화탄소가 궁극적으로 환원되는 것으로 예상되고 있으나, 각 Re 화합물의 중간체 구조 등은 아직까지 확인된 바 없다.
3.1.2. 이성분계(two component system)
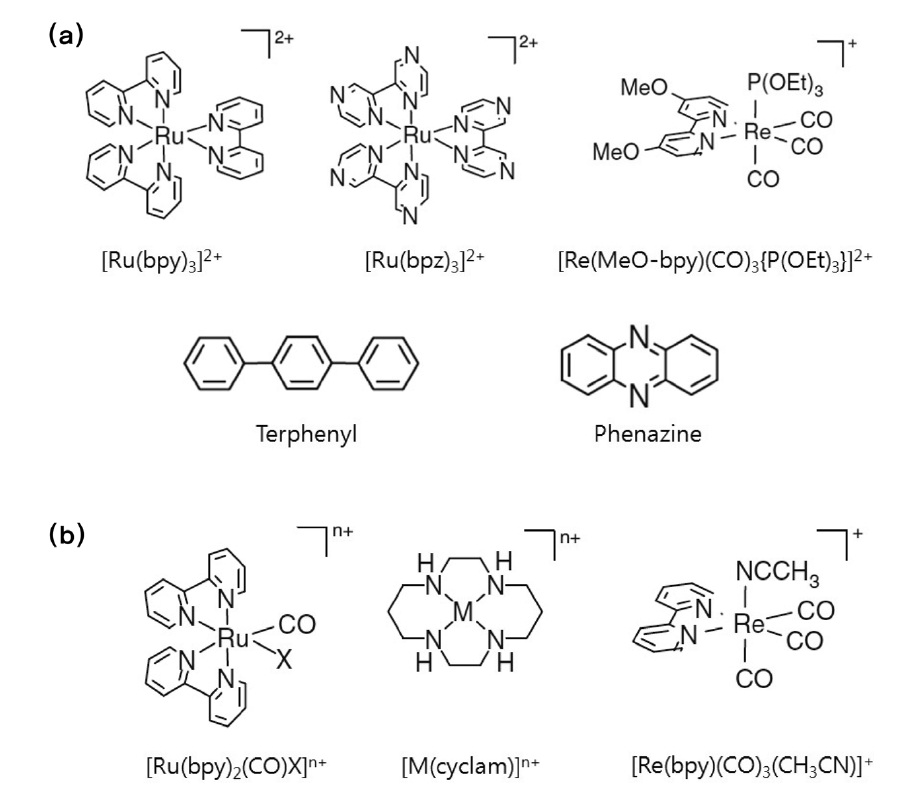
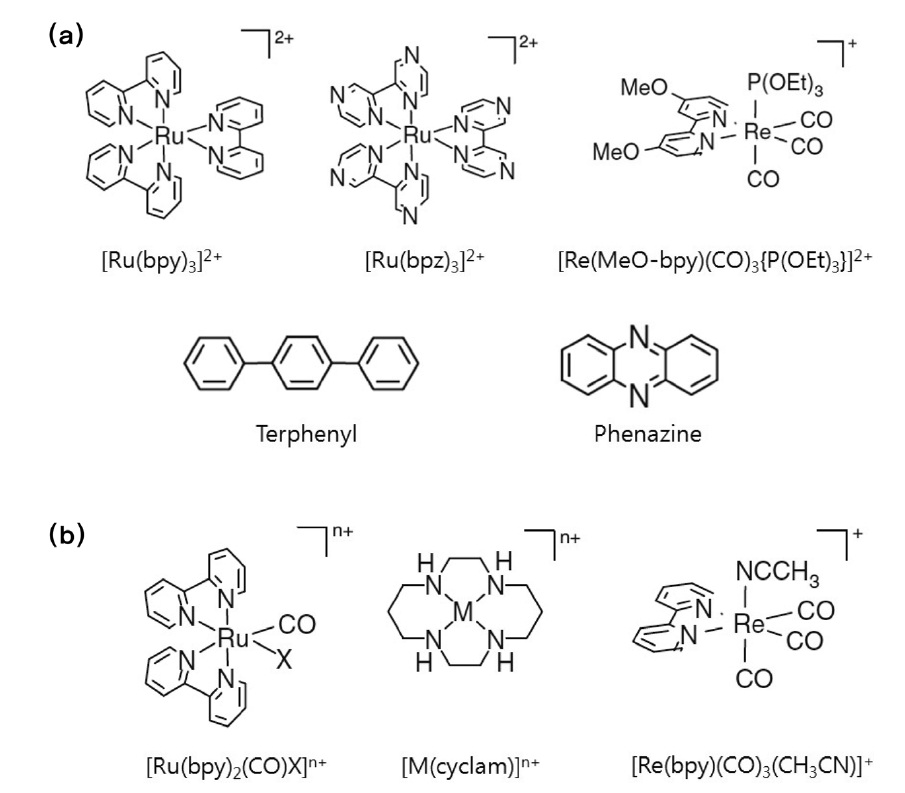
이성분계 분자 촉매는 광감응제 기능과 촉매 기능을 담당 하는 두 개의 다른 분자가 결합되어 있는 구조이며, 이성분계에서의 이산화탄소 전환반응은 식 (31)~(33)에서처럼 빛에너 지에 의해 여기된 광감응제 분자(P+)가 용액 상에 첨가된 전자 공여체(electron donor, D)에 의해 환원된 형태의 분자(P-)로 전환된 후, 촉매 분자에 받은 전자를 전달하게 되면서 촉매 사이트가 활성화되는 메커니즘으로 간략히 설명할 수 있다. 몇 가지 대표적인 광감응제 분자와 촉매 분자의 구조는 Figure 8에서 확인할 수 있다. 본문은 광감응제 분자를 기준으로 정리 하였다.

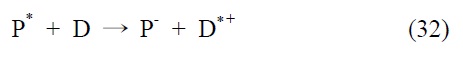
3.1.2.1. Ru 광감응제
Ru 화합물은 현재 가장 널리 사용되고 있는 대표적인 광감 응제 분자이다. 가시광을 효과적으로 흡수할 수 있고 대략 1 μs 수준의 상대적으로 긴 시간동안 여기된 상태로 존재할 수 있
기 때문이다. Ru 화합물 가운데 가장 많이 사용되고 있는 광감 응제 분자는 [Ru(bpy)3]2+ (bpy = bipyridine)이며, [Ru(bpz)3]2+ (bpz = bipyrazine)이 때때로 사용되기도 한다. Willner et al.[50] 은 [Ru(bpy)3]2+ 을 광감응제로 사용하고 Ru이나 Os 콜로이드를 촉매로 사용하였는데, 용액상에 트리에탄올아민 전자공여체(triethanolamine electron donor)와 바이피리디니움 전자전달 체(bipyridinium electron relay)를 첨가하여 이산화탄소의 가시광 조사하에서 메탄과 수소 등으로 환원되는 것을 확인하였다. 이들은 [Ru(bpz)3]2+ 광감응제/Ru 콜로이드 촉매 시스템을 사용했을 경우에는 메탄이 생성된 반면 수소는 발생하지 않는 것도 관찰하였다[51].
Ishida et al.[52-54]은 [Ru(bpy)2(CO)2]2+ 촉매를 이용하여 전기화학적으로 이산화탄소를 성공적으로 환원시키는 결과를 확인한 후, 이 촉매에 [Ru(bpy)3]2+ 광감응제를 첨가하여 이산화탄소의 광화학적 환원 반응을 유도한 결과 주요 부산물로 일산화탄소와 개미산을 얻었다. 이후 Lehn and Ziessel[55]에 의해서 비슷한 형태의 [Ru(bpy)2(CO)X]n+ (X = Cl, H, n = 1 or X=CO, n = 2) 균일계 촉매를 연구하여 이 가운데 [[Ru(bpy)3]2+ 광감응제와 [Ru(bpy)2(CO)H]+ 촉매 시스템에서 15&의 최대 광효율로 이산화탄소가 개미산으로 전환됨을 보고하였다.
또한 [Ru(bpy)3]2+ 광감응제와 함께 Tetraaza-macrocyclic 화 합물이 촉매로 사용되기도 하였는데, Tinnenman et al.[56]은 Ni과 Co tetraaza-macrocyclic 화합물을 촉매로 이용하여 광화 학적/광전기화학적으로 이산화탄소를 전환시켜 수소와 함께 일산화탄소를 주요 부산물로 얻었고, Grant et al.[57] 역시 같 은 Ru 광감응제에 [Ni(cyclam)]2+ 촉매를 이용하여 아스코르빈산(ascorbic acid) 전자공여체 존재하에 이산화탄소가 일산화탄소로 전환되는 것을 확인하였다. 또한 이들은 14C로 표지된 CO2 를 이용하여 CO가 CO2로부터 직접 전환된 것임을 증명하기도 하였다. Ru 광감응제와 Ni (cyclam) 촉매분자 혹은 Re 화합물 촉매분자가 화학적으로 결합된 거대분자 화합물(supramolecular complex)이 합성되기도 하였는데, 거대분자 화합물의 경우 이 분자계(bimolecular system)보다 더 안정적인 형태의 화합물로 존재할 뿐 아니라 이산화탄소 환원반응에 대한 촉매 활성도 더 뛰어난 것으로 보고되었다[58-60]. 이는 두 분자가 화학적으로 결합되어진 거대분자 화합물의 경우 광감응제 분자의 여기에 의해 생성된 전자가 촉매 분자로 이동할 때 분자 내 전자 전달이 가능하기 때문일 것으로 추정된다.
3.1.2.2. Re 광감응제
Re 화합물은 앞서 살펴보았듯이 그 자체로 광감응제와 촉매 역할을 모두 담당할 수 있기 때문에 단일성분계 물질로 분류되었다. 그러나 광감응제 및 촉매로서의 활성이 각기 다른 두 종류의 Re 화합물을 함께 반응시킴으로 인해 이산화탄소의 전환 효율을 높이는 이성분계 반응도 시도되었다. Takeda et al.[46]은 다양한 Re 화합물을 각각 광감응제와 촉매로 활용하여 이산화탄소의 광화학적 환원반응을 진행한 결과 [Re {4,4'-(MeO)2bpy}(CO)3{P(OEt)3}]+ 광감응제와 [Re(bpy)(CO)3 (CH3CN)]+ 촉매 이성분계 반응에서 이산화탄소가 일산화탄 소로 환원되며, 광효율은 약 59%에 이르는 것으로 보고하였 다. 이는 알려진 균일계 촉매 가운데 가장 효율적인 시스템이 라 할 수 있다. 그러나, Re 화합물을 광감응제로 활용할 경우, Ru 화합물과 달리 지표에 도달하는 태양광의 대부분을 차지하고 있는 가시광 영역을 흡수할 수 없다는 점이 문제로 지적되고 있다.
3.1.2.3. 터페닐 및 페나진 광감응제
터페닐(terphenyl)과 페나진(phenazine) 분자를 광감응제로 활용한 연구도 보고되었는데 이 경우에는 앞서 언급되었던 Co (cyclam) 계열의 촉매가 함께 이용되었다[61,62]. 터페닐 광 감응제의 경우, Co (cyclam)을 포함한 8개의 tetraaza-macrocyclic 화합물을 촉매로 선택하였고 트리에틸아민 혹은 트리에탄올아민을 전자공여체로 첨가하여 이산화탄소의 환원 반응을 유도한 결과 일산화탄소, 개미산이 부산물로 생성되고 아울러 부가반응으로 수소도 함께 생성되는 것으로 드러난 반면, 페나진 광감응제의 경우에는 개미산이 주요 부산물로 발생하며 일산화탄소 및 수소의 발생량이 극히 적은 것으로 나타났다.
3.2. 비균일계 반응 시스템(heterogeneous reaction system)
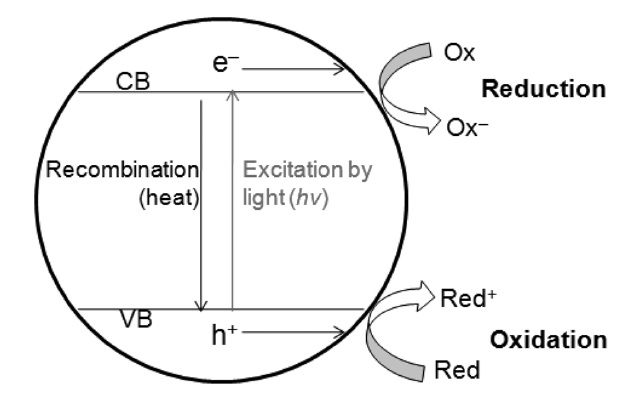
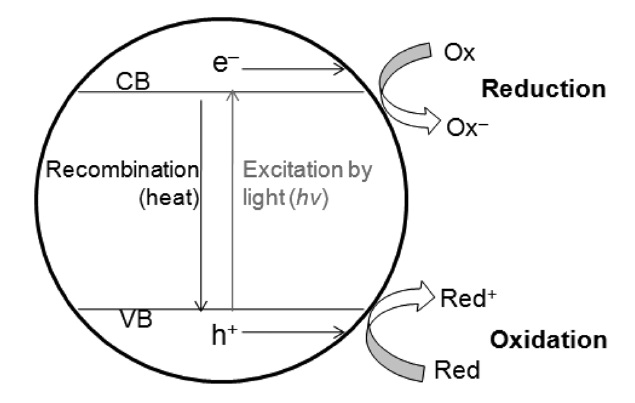
비균일계에서의 광화학적 이산화탄소 환원반응은 무기 반도체 광촉매(inorganic semiconductor photocatalyst)에 의해서 유발된다. 반도체 광촉매라 함은 밴드갭(band gap) 에너지에 해당하거나 그보다 큰 빛에너지를 조사할 경우 여러 가지 광 화학적 반응이 유발되는 반도체 특성을 갖는 물질을 통칭한
다. 반도체 광촉매의 원리는, Figure 9에서 보는 것과 같이 전자가 채워져있는 광촉매의 가전자대(valence band, VB)의 전자가 빛에너지를 흡수하며 전자가 비어있는 전도대(conduction band, CB)로 여기하게 되고, 이에 가전자대에서는 전자가 빠져나간 정공(hole)이, 전도대에서는 여기된 전자(excited electron) 가 각각 생성되는데, 이때 생성된 전자와 정공의 환원력과 산화력을 이용하여 다양한 산화환원 반응을 유도할 수 있게 된다. 무기 광촉매를 사용한 이산화탄소의 광화학적 전환 반응은 전도대의 여기된 전자를 이용하여 이산화탄소를 환원 시키는 반응이며 이때 사용학 광촉매의 전도대 위치가 이산화탄소의 환원전위보다 높아야만 이산화탄소의 환원반응이 열역학적으로 가능하게 된다. 여러 광촉매 물질 가운데 이산화티탄(TiO2)을 이용한 이산화탄소의 광화학적 전환 연구가 가장 활발하게 진행되고 있는데 이는 이산화티탄이 경제성, 화학적 안정성, 무독성, 그리고 여러 다른 광반응에서 높은 광활성을 보이는 등의 장점을 가지고 있기 때문이다. 따라서 본 단락에 서는 이산화티탄 광촉매를 이용한 이산화탄소의 광화학적 전환 연구와 기타 무기광촉매 물질들을 활용한 전환 연구로 구분하여 정리하였다.
3.2.1. 이산화티탄 광촉매(titanium dioxide photocatalysts)
TiO2 및 기타 반도체 광촉매 물질을 이용한 수용액상에서의 이산화탄소 환원반응은 Inoue et al.[63]에 의해서 처음으로 알려졌는데 이때 주요 환원 부산물로는 메탄올이 발생하였고, 그외 개미산과 포름알데히드 등도 함께 생성되었다. 이는 균일계 촉매의 경우 주요 부산물이 일산화탄소 혹은 개미산에 그치 는 것에 반해 비균일계 무기 광촉매 시스템은 다전자 전달 반응에 더 유리함을 보여주고 있다. 따라서 현재까지 개발된 광촉매 시스템 상에서 이산화탄소를 메탄올이나 메탄 등의 부가 가치를 갖는 화학연료로 선택적으로 전환하는 반응에 있어서는 무기 광촉매계열이 더 적합하다고 볼 수 있겠다. 그러나 조사된 광량 대비 이산화탄소의 전환율이 메탄올 생성을 기준으로 0.02%, 포름알데히드 생성 기준으로 0.05%에 불과해 매우 낮은 효율을 보였다. 이후 다양하게 개질된 TiO2를 중심으로 한 광화학적 이산화탄소 전환 연구가 꾸준히 진행되어져 오고 있으며, 주요 부산물로 일산화탄소, 개미산, 포름알데히드, 메탄올, 메탄, 에틸렌 등이 생성되었다[64-67].
광촉매 시스템에서는 여기된 전자와 정공의 재결합을 방지함으로써 보다 효율적인 환원 반응을 유도할 수 있기 때문에 정공에 의해 유발되는 산화반응 역시 중요하게 살펴보아야 한다. 비균일계 촉매를 이용한 이산화탄소의 환원반응은 주로 수용 액상에서 진행되므로 이 경우 가전자대에서는 물의 산화반응 (water oxidation)이 일어나게 되는데 물의 산화반응이 전체 반응의 속도결정단계(rate determining step)일 가능성이 높기 때문에, 이에 대한 해결방안으로 용액상에 프로판올과 같은 알콜 계 전자공여체를 첨가하여 빠른 산화반응을 유도함으로써 이산화탄소의 환원반응을 효과적으로 진행시키는 방법이 널리 사용되고 있다[68-70]. Kaneco et al.[69,71]은 프로판올 용액 상에서 TiO2광촉매를 이용하여 이산화탄소를 2.8 MPa 고압 조건하에서 광화학적으로 환원시키면 메탄이 선택적으로 생성되는 것을 확인하였고, 또한 반응 압력을 6.5 MPa로 더 높여 액체상의 CO2 상태에서 반응을 진행하면서 물을 첨가할 경우 에는 개미산이 선택적으로 생성됨을 보고하였다. 이는 사용되는 전자공여체와 양성자 제공물질에 따라 이산화탄소의 환원 부산물이 달라질 수 있음을 보여준다.
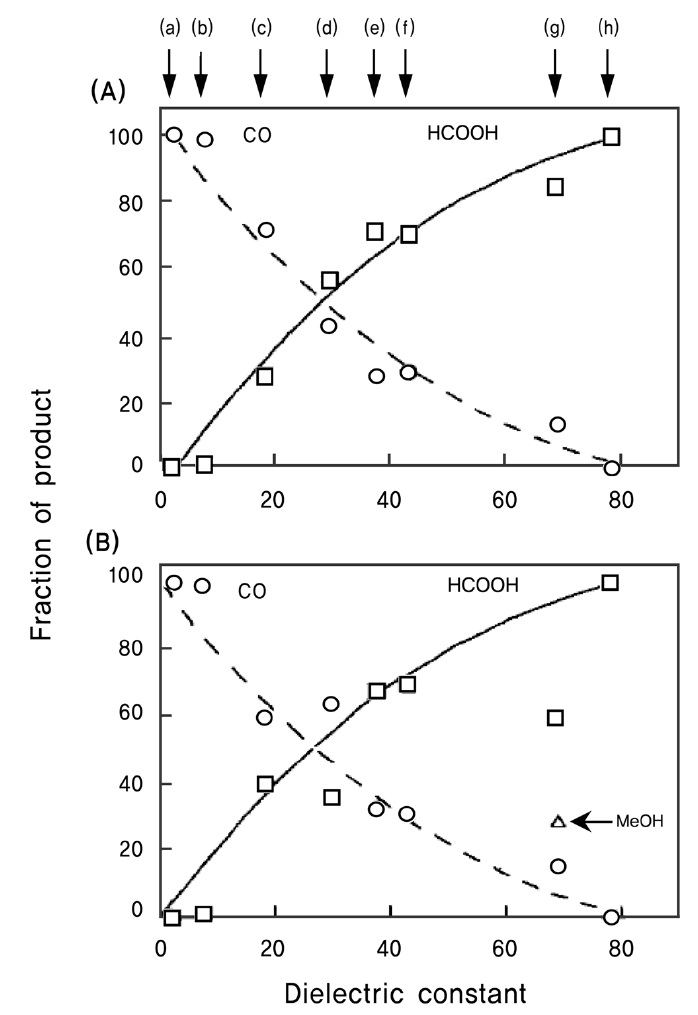
한편, 수용액 반응의 경우 이산화탄소의 물에 대한 용해도가 매우 낮고, 이산화탄소의 환원반응이 물분해 수소생성 반응과 경쟁을 하게 되는 등의 한계로 인하여 유기용매 상에서의 이 산화탄소 환원 반응도 아울러 연구되어졌다. Liu et al.[72]은 TiO2/SiO2 광촉매를 이용한 이산화탄소 환원반응에서 물, 아세토니트릴, 프로판올, 디클로로메탄 등 유전상수가 다른 여러 가지 용매를 사용한 결과, Figure 10에서 보는 것과 같이 유전 상수가 큰 용매를 사용할수록 개미산의 생성은 증가되고 일산화탄소의 생성비율은 감소함을 규명하였다. 또한 Tseng et al.[73]은 수산화나트륨을 첨가할수록 이산화탄소의 메탄올 전환 효율이 높아지는 것이 관찰되었는데, 이는 수산화나트륨을 첨가함으로써 이산화탄소의 용해도가 증가되고 첨가된 OH- 가 전자공여체로 활용되어 전자-정공의 재결합 반응을 늦출 수 있었기 때문인 것으로 사료된다.
아울러 여러가지 금속 혹은 금속산화물을 조촉매(co-catalyst) 로 사용하여 이산화탄소 환원 효율의 향상 및 환원생성물의 선택성을 증가시킬 수 있다. 주로 구리(Cu), 루테늄(Ru), 팔라 듐(Pd) 등이 이산화탄소 환원을 위한 조촉매로 활용되었는데, 이 가운데 구리 계열 조촉매는 메탄 및 메탄올 등으로의 선택적 환원에 큰 효과를 가진 것으로 알려져 있다. Adachi et al.[74] 은 아나타제(anatase) 결정 구조를 가지고 있는 TiO2 표면에 5 wt% 미만의 구리를 도핑할 경우 이산화탄소가 메탄과 에틸렌으로 환원되는 것을 보였는데, 이때 구리의 비율이 5 wt% 를 넘어갈 경우 주된 반응이 이산화탄소 환원반응이 아닌 물분해 수소 생성 반응으로 전환되는 것으로 나타났다. 또한 Ishitani et al.[75]는 다양한 금속을 조촉매로 사용하였을 경우 이산화탄소의 환원 효율을 비교하였는데, 비교된 6개의 금속들 가운데 팔라듐(Pd)이 도핑된 이산화티탄 촉매가 이산화탄소의 메탄으로의 전환 반응을 크게 향상시키는 것으로 나타났다. 한편, 특정 조건하에서 Cu 혹은 CuO가 도핑된 이산화티
탄을 이용할 경우 메탄올이 선택적으로 생성되는 것도 보고되었다[73,76].
TiO2 광촉매와 제올라이트 혹은 다공성 실리카와 결합된 형태의 촉매도 사용되었는데, 이 경우 역시 메탄이나 메탄올 등이 주요 이산화탄소 환원생성물로 검출되었으며 이산화티탄만 사용한 경우보다 이산화탄소의 환원 효율이 더 높은 것으로 나타났다[77-79]. 이는 TiO2 나노 입자들이 다공성 구조체 안에서 보다 잘 분산된 형태로 생성되거나 기공에 따라 TiO2의 사이즈나 결정성 등이 달라지기 때문일 것으로 보여진다. 아울러 Xia et al.[80]은 이산화티탄을 다중벽 탄소나노튜브(multiwalled carbon nanotube, MWCNTs)에 담지시킨 촉매를 합성하여 이산화탄소 전환 효율을 현저하게 향상시켰는데, 특이한 것은 이들이 MWCNTs 상에서의 이산화티탄 합성법에 따라 환원 부산물이 각기 다르게 생성되었다. 즉, 졸겔법(sol-gel method)으로 합성된 경우에는 메탄올이, 수열법(hydrothermal method)으로 합성된 경우에는 개미산이 생성되었다.
Kim et al.[81]은 최근에 Pd 조촉매를 도핑한 이산화티탄 표면을 나피온(nafion)으로 코팅하여 자외선 조사 하에 전자공여체의 첨가 없이 이산화탄소를 메탄이나 에탄 등으로 환원시킬 수 있음을 보고하였다. 이는 나피온 코팅을 통해 이산화티탄 표면에서의 양성자 활동도를 증가시킴으로 인해 양성자가 연계된 다전자전달 반응(proton coupled multielectron transfer, PCET)이 용이해지고, 생성된 부산물의 재산화가 방지되기 때문인 것으로 사료된다.
3.2.2. 기타 무기 광촉매(other inorganic photocatalysts)
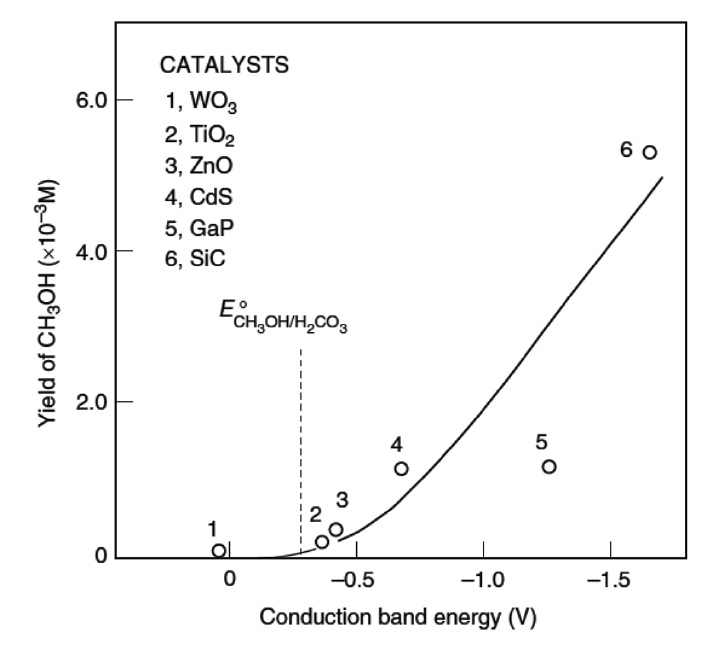
이산화티탄 외에도 기타 광촉매 물질들이 이산화탄소의 광화학 전환에 응용되었다. Figure 11은 다양한 광촉매 물질의 이산화탄소 전환반응을 보여주고 있는데, 사용된 광촉매 물질의 전도대 전위가 더 낮을수록 메탄올이 더 많이 생성되는 경향성을 있음을 확인할 수 있다[63].
그 외에도 MgO, ZrO2, Ga2O3, ATaO3 (A = Li, Na, K) 등의 금속 산화물 광촉매를 이용한 이산화탄소 환원 반응이 보고 되었고, 이 가운데 특별히 상대적으로 밴드갭 에너지가 작은 InTaO4, BiVO4 등의 광촉매를 이용했을 경우 가시광 조사 하에서도 이산화탄소 환원 반응이 진행될 수 있는 것으로 나타났다[82-87]. Liu et al.[86]은 마이크로웨이브를 이용한 수열합 성법을 통해 400~500 nm 파장의 빛에너지를 흡수할 수 있는 BiVO4 촉매를 합성하여, 가시광 조사하에 에탄올이 선택적으로 생성됨을 확인하였다. 또한 Pan and Chen[87]은 2.7 eV의 밴드갭 에너지를 가진 InTaO4 광촉매와 NiO를 조촉매를 사용하여 KHCO3 수용액상에서 반응을 진행시킨 결과 가시광 조사 하에 메탄올이 생성되는 것을 확인하였다.
금속 산화물 외에도 CdS, ZnS, CdS-ZnS 등의 칼코겐 화합
물도 이산화탄소 환원 광촉매로 활용되었다[88-91]. CdS 광촉매의 경우, CdS 나노입자를 다양한 용매상에서 합성하여 이산화탄소 환원 반응에 이용하였는데 이때 여분의 Cd 이온이 존재할 경우 그 광활성이 현저하게 증가하는 것으로 나타났고, 용매는 dimethylformamide를 사용했을 경우가 가장 효과적인 것으로 나타났다[88,89]. 이 때 주요 환원 부산물은 일산화탄소 인 것으로 밝혀져 이산화티탄 광촉매를 사용했을 경우와 대조를 이루었다. 아울러 합성에 사용된 Cd 전구물질(precursor)과 반응에 사용된 전자공여물질에 따라 이산화탄소의 전환 효율이 달라짐도 확인되었는데, Cd(CH3COO)2 전구체와 트리에틸아민 전자공여체를 사용했을 경우에 가장 효율이 높은 것으로 드러났다[89]. ZnS의 경우, Inoue et al.[91]이 여러 가지 금속 조촉매가 도핑된 ZnS와 CdxZn1-xS로 표현되는 CdS-ZnS 나노입자 혼성 촉매를 합성하여 이산화탄소 전환에 응용하였는데, 순수한 ZnS의 경우 개미산의 주요 환원생성물이었으나 CdS와의 혼성비율이 증가될수록 개미산의 생성은 줄어들고 일산화탄소와 수소의 생성이 증가하는 것을 확인할 수 있다. 그러나 카드뮴이 금속 조촉매로 ZnS 표면에 존재하게 될 때에는 이산화탄소 환원 광활성 및 개미산의 생성을 증가시키는 것으로 나타났다.
최근 들어 TiO2 비균일계 광촉매에 균일계 광촉매나 광감응제를 혼성시키거나 기타 다른 기능을 가진 물질들과 혼합한 혼성 시스템(hybrid system)이 많이 연구되어지고 있는데, 이 혼성 시스템을 이용한 이산화탄소의 광화학적 환원 연구 도 몇몇 연구진에 의해 시도되었다. 이산화티탄 혼성 시스템의 경우, 이산화티탄의 밴드갭 에너지가 3.2 eV로, 오로지 자외선 영역에서만 광활성을 보인다는 단점을 가지고 있기 때문에, 가시광을 흡수할 수 있는 광감응제 물질와 결합한 시스템이 많이 보고되고 있다. 이때 광감응제로 사용되는 물질은 주로 균일계 촉매시스템에서 널리 사용되고 있는 Ru 화합물 이나 CdSe와 같이 밴드갭 에너지가 작은 비균일계 무기 광촉매이다[66,92,93]. 이같은 혼성시스템에서는 밴드갭이 작은 비균일계 광촉매나 Ru 광감응제 물질이 빛을 흡수하여 여기된 전자가 이산화티탄의 전도대로 이동하여 이산화티탄 표면에서 이산화탄소의 환원반응이 일어나게 된다. CdSe의 경우, Bulk CdSe-TiO2 간의 전자 전달이 불가능하나 CdSe 나노입자를 사용하며 CdSe 전도대의 위치가 높아져(negatively shifted) TiO2으로 전자를 주입할 수 있는 것으로 알려졌다. Wang et al.[92]은 나노입자의 CdSe/Pt/TiO2 혼성 시스템을 이용하여 가시광 조사 하에 수용액상에서의 이산화탄소 환원반응을 유발하여 수소나 일산화탄소의 발생 없이 메탄과 메탄올로 선택적으로 환원이 가능함을 보고하였다. 또한, Ozcan et al.[93]은 [Ru(bpy)3]2 /Pt/TiO2 혼성시스템으로 이산화탄소를 메탄으로 전화시켰다. 한편, Woolerton et al.[94]은 Ru 화합물과 TiO2 나노입자가 결합된 비균일계 시스템에
3.4. 광전기화학 반응 시스템(phtoelectrochemical reaction system)
무기 반도체 광촉매 물질을 전극으로 사용할 경우, 광에너지뿐 아니라 전기에너지를 동시에 에너지원으로 사용하여 이산화탄소를 전환시키는 광전기화학 반응(photoelectrochemical reaction)도 가능하게 된다. 즉, 전기화학적 이산화탄소 전환 반응 시스템에서 사용하는 전극을 무기 반도체 광촉매 물질로 사용할 경우, 전기화학적 반응과 동시에 반도체 광촉매 물질에 의한 광화학적 반응도 진행될 수 있는 광전기화학 시스템이 가능해진다. 반도체 광전극을 이용한 광전기화학적 시스템을 이산화탄소 환원 반응에 적용하려면, 조사된 전극 표면에서 환원반응을 일으킬 수 있는, 즉 전자는 고체-액체 계면으로, 정공은 벌크로 이동시키는 p형 반도체 전극을 사용해야만 한 다. 현재까지 InP, GaP, GaAs, CdTe 등의 반도체 물질들이 광전기화학적 이산화탄소 환원 반응에 광전극으로 사용되었다. 이 때 상대전극으로는 주로 금속 전극이 사용되며 간혹 광산 화반응이 가능한 n형 반도체 전극이 사용되기도 한다. 본 단락에서는 광전기화학적 이산화탄소 환원 반응에 사용된 반도체 광전극을 중심으로 1) 반도체 광전극을 그대로 사용한 시스템, 2) 전극 표면을 개질한 시스템, 그리고 3) 반도체 전극과 균일계 분자 촉매를 함께 사용한 혼성 시스템으로 나누어 정리 하였다.
3.4.1. 반도체 전극을 이용한 광전기화학 시스템
반도체 전극을 이용한 광전기화학적 이산화탄소 환원반응은 Halmann[95]에 의해 처음으로 보고되었다. 실험에서 p-GaP 전극을 사용하여 -1.0 V(칼로멜 전극기준)의 전압을 걸어줌과 동시에 빛을 조사한 결과 수용액상에서 이산화탄소가 개미산 으로 전환되는 것을 확인하였다. Inoue et al.[96]역시 p-GaP 전극을 사용하여 -1.5 V (vs. SCE)의 전압을 걸어주며 빛을 조사한 결과 0.5 M H2SO4 수용액 상에서 개미산과 포름알데히드, 메탄올 등이 생성되는 결과를 보고하였다. 그러나 GaP의 경우 밴드갭 에너지가 2.3 eV 정도로 다른 반도체 전극에 비해 상대적으로 높은 편이기 때문에 이에 밴드갭 에너지가 작은 반도체 물질을 광전극으로 사용하는 연구도 시도되었는데, Taniguchi et al.[97]은 밴드갭 에너지가 1.4 eV 정도인 p-CdTe 전극을 이용하여 600 nm 장파장의 빛을 조사함과 동시에 -1.6 V (vs. SCE) 전압을 걸어주어 70%에 달하는 전환효율(faradaic efficiency)로 일산화탄소가 생성됨을 확인하였다. 이 경우, 순수한 수용액상이 아닌, 5% 물과 DMF 혼합 용액이 사용되었고 tetrabutylammonium perchlorate (TBAP)가 전해질로 사용되었 는데, tetra-alkylamoonium 염을 사용할 경우 이산화탄소의 환 원 효율이 증가하는 것으로 나타났다. 아울러 p-GaAs (EBG = 1.4 eV)와 p-InP (EBG = 1.3 eV ) 등의 반도체 광전극도 이용되 었는데, Canfield and Frese[98]은 수용액 상에서 각각의 전극을 사용하여 50~80% 수준의 높은 전환효율로 메탄올이 생성됨을 보고하였다. 그러나 이 경우에도 마찬가지로 -1.2~-1.4 V (vs. SCE)의 상대적으로 매우 큰 전압이 요구되었는데 이는 위의 반도체 광전극을 그대로 사용할 경우 빛에 의해 시스템이 활성화되기는 하나 실제적인 이산화탄소의 전환 반응은 외부에서 공급해주는 전기에너지에 의해 유발되기 때문일 것 이다. 따라서 언급된 광전극을 사용한 광전기화학적 시스템은 전기화학적 이산화탄소 전환 반응과 비교할 경우 전극차 이에 따른 물질적 특성 외에는 큰 장점을 찾기 어렵다고 볼 수 있다.
3.4.2. 개질된 반도체 전극을 이용한 광전기화학 시스템
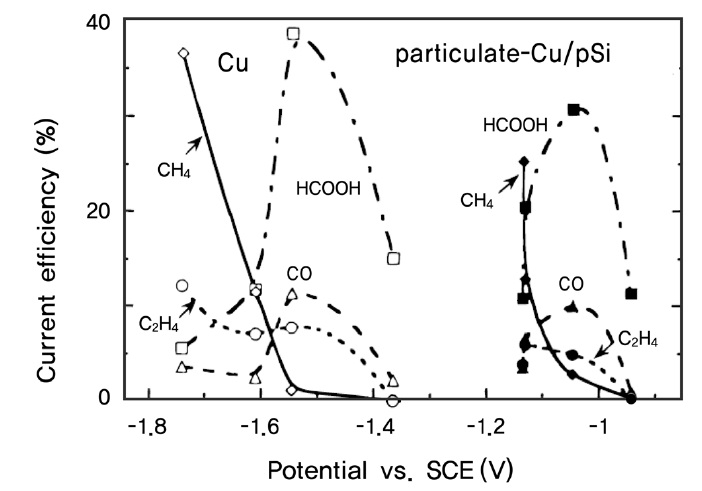
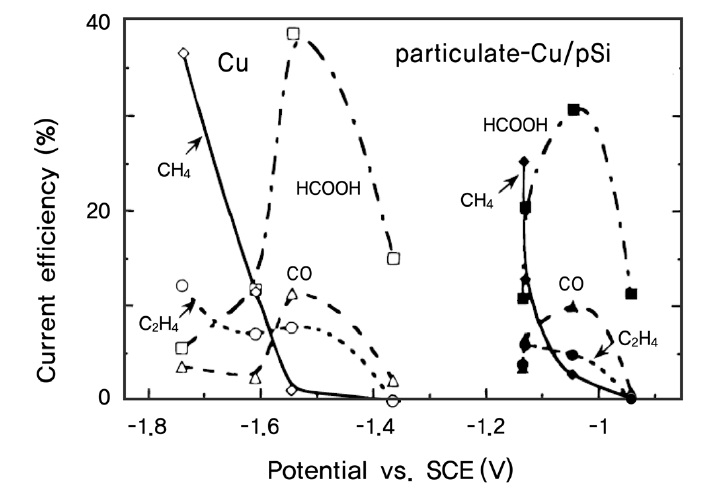
반도체 전극에 여러 가지 촉매적 활성을 가진 물질을 더하여 이산화탄소의 환원효율을 증가시키거나 환원 반응의 개시 전위(onset potential)를 낮추기 위한 노력도 진행 중인데, 주로 위에서 사용된 반도체 광전극 표면에 여러 가지 금속들을 증 착시키는 방법을 택하고 있다. Ikeda et al.[99,100]은 p-GaP 에 Pb, Zn, Au, Cu 등의 금속을 증착시킨 결과, -1.25 V (vs. SCE) 수용액상에서 Pb, Zn를 함유된 전극에서 이산화탄소의 일산화탄소 및 개미산으로의 전환효율이 증가하고, Cu가 함유된 전극에서는 -1.6 V (vs. SCE) 전압하에서 메탄으로의 전환효율이 증가함을 보였다. Hinogami et al.[101]은 p-Si 전극 을 Cu, Ag, Au 등의 금속 나노입자로 코팅하여 사용할 경우 Figure 12에서 보는 것과 같이 금속 전극을 사용할 경우보다 이산화탄소의 환원반응을 유발할 수 있는 개시전위가 낮아짐을 관찰하였다. 이때 가해주는 전압에 따라 생성되는 이산화 탄소 환원 부산물의 조성이 달라지는 것도 관찰되었는데, 상대적으로 낮은 전위에서는 일산화탄소와 개미산이 생성되나 -1.5 V (Cu/p-Si 전극의 경우 -1.0 V) 이상의 높은 전위에서는 메탄과 에틸렌의 발생량이 증가하는 것으로 나타났다. 한편 Kaneco et al.[102]은 p-InP 표면을 다양한 금속 나노입자로 개질하여 LiOH/Methanol 전해질 용액 상에서 광전기화학적 이산화탄소의 환원 반응을 살펴보았는데, 도핑된 금속 입자
에 따라 환원 부산물의 조성이 달라지는 것으로 나타났다. 즉, Pb, Au, Ag, Cu 등이 도핑된 경우 일산화탄소와 개미산이 생성되나 Pd를 도핑하면 일산화탄소만 생성되고, Ni/p-InP에서 는 일산화탄소, 개미산과 함께 메탄과 에틸렌도 아울러 생성 되었다.
3.4.3. 균일계 분자촉매와 반도체 전극을 이용한 혼성 광 전기화학 시스템
반도체 광전극을 사용하여 광전기화학적 이산화탄소 환원 반응을 유도할 경우, 반도체 광전극을 안정화시키고 효율을 증가시킬 수 있는 다른 방법 중의 하나가 균일계 분자 촉매, 즉 용액 상에 반도체 전극에서 효과적으로 전자를 받아 이산화탄소를 환원시킬 수 있는 매개체(solution mediater)를 첨가하는 방법이다. Taniguchi et al.[103]은 15-crown-5-ether 유기 매개체를 용액 상에 첨가하여 p-GaP 광전극 시스템에서 메탄올, 개미산, 포름알데히드 등이 높은 전환효율로 생성되는 것을 확인하였고, 이와 유사하게 Bockris and Was[104]s는 p- CdTe 광전극과 crown ether 유기매개체를 이용한 이산화탄소 환원 반응을 보고하였다. 한편, Parkinson and Weaver[105]는 p-InP 광전극 시스템에 메틸 바이올로젠(methyl viologen) 전자전달체와 개미산 탈수소화 효소를 첨가하여 수용액상에서 -0.2 V (vs. SCE)의 낮은 전압만을 가하여 이산화탄소를 개미산으로 효율적으로 전환하는 데 성공하였다.
아울러 기타 균일계 분자촉매를 매개체로 사용하는 광전기 화학 시스템도 보고되었다. Bradley and Tysak[106]은 tetraazomacrocyclic metal 화합물과 p-Si 광전극의 접합을 통해 광조사하에 p-Si 광전극 표면에서 금속 테트라아조 거대분자 화합물(tetraazomacrocyclic metal complex)이 환원되고, 환원된 분자 촉매가 이산화탄소를 환원시켜 일산화탄소를 생성함을 보여주었다. 또한 Beley et al.[107]과 Petit et al.[108]은 각각 p- GaAs, p-GaP 광전극 시스템에서 Ni(cyclam)2+ 균일계 분자촉매를 수용액상에 첨가하여 -0.10 V 또는 -0.44 V (vs. SCE)의 상대적으로 낮은 전압 하에 일산화탄소로의 전환효율을 100% 가까이 달성하였다. 또한 Re(bpy)(CO)3Cl 분자촉매와 p-Si 혼성 시스템, 피리디니움(pyridinium) 분자촉매와 p-GaP 혼성 시스템 등도 보고되었다[109,110].
추가적으로 이러한 분자촉매를 용액 상에서 매개체로 사용 하는 것이 아니라 광전극 표면에 결합, 고정시키는 방법도 연 구되었는데, 이는 고정된 시스템의 경우 용액상에서 균일계 촉매의 침전 등을 문제를 방지할 수 있고, 동시에 보다 효율 적인 전자전달이 가능해져 효율을 증가시키는 등의 장점을 가 지고 있으며 또한 물에 녹지 않는 분자 촉매들도 수용액상에 서 사용할 수 있기 때문이다. 예를 들어 Cabrera and Abruna [111]은 Re(CO)3(4-vinyl,4'-methyl-2,2'-bipyridine)Cl 분자 촉 매를 p-Si, p-WSe2 광전극에 고정시킨 혼성 시스템을, Arai et al.[112]은 Ru 폴리머 화합물([Ru(L-L)(CO)2]n, L-L = diimine ligand) 분자촉매를 p-InP 광전극에 고정한 혼성 시스템을 구축하여 효율적인 광전기화학적 이산화탄소 환원 반응을 유도하였다. 이러한 분자 촉매가 광전극에 고정된 혼성시스템의 경우 분자촉매와 광전극간의 직접적 결합을 만들기 어려운 관 계로 폴리머를 이용한 결합이 주로 사용되고 있는데, 앞으로 직접적 결합을 통한 혼성 광전기화학 시스템의 개발이 요구된 다고 볼 수 있다.
광화학적 이산화탄소의 전환에서는 금속 화합물 분자촉매를 포함한 균일계와 무기 반도체 광촉매를 포함한 비균일계 촉매를 중심으로 활발히 연구가 진행되고 있다. 균일계 촉매의 경우 [Ru(bpy)3]2+ [Ru(bpy)2(CO)2]2+ 등의 Ru 화합물과 Re(CO)3 (bpy)X 형태의 Re 화합물이 대표적인 광감응제 혹은 균일계 촉매로 사용되고 있으며, 금속과 결합된 포피린 화합물 등의 금속 거대고리분자 촉매도 광화학적 이산화탄소 환원 반응을 일으키는 것으로 알려졌다. 균일계 분자촉매는 흡광계수가 매우 높아 광에너지를 효율적으로 포획하여 반응에 참여시킬 수 있기 때문에 비교적 우수한 광전환 효율을 보이고 있으나, 현재까지 알려진 촉매의 경우 이산화탄소의 주된 환원 부산물로 일산화탄소 혹은 개미산을 생성하기 때문에 더 많은 전자의 전달이 요구되는 메탄과 메탄올 등의 부가가치를 가진 부산물로의 선택적 환원 반응은 불가능하다. 또한 분자 촉매 자체의 광분해 가능성 및 유기 분자촉매의 경우 수용액 상에서 사용할 수 없다는 등의 문제점도 앞으로 더 고려되어야 할 부분으로 생각된다. 한편 비균일계 촉매 시스템은 TiO2 반도체 광 촉매를 중심으로 많은 연구가 진행되어져 왔는데, TiO2는 경제성, 화학적 안정성 및 광활성 등의 측면에서 매우 뛰어난 장점을 가지고 있는 광촉매로, 광화학적 이산화탄소 전환반응에 적용할 경우 일산화탄소나 개미산 뿐만 아니라 메탄올, 메탄, 에탄올, 에틸렌 등의 다전자전달 반응에 의한 환원 부산물도 생성되기 때문에 그 가능성을 인정받고 있다. 그러나 아직까지 조사된 광에너지 대비 생성된 이산화탄소 환원 부산물의 비율을 나타내는 광효율이 매우 낮기 때문에 보다 효율적인 촉매 시스템의 개발이 시급히 요구되고 있다. 또한 TiO2 경우, 3.2 eV의 비교적 높은 밴드갭 에너지를 가지고 있기 때문에 태양광의 대부분의 영역을 차지하고 있는 가시광 영역을 활용할 수 단점을 가지고 있어 분자 광감응제 및 밴드갭 에너지가 작은 기타 광촉매 등과 결합시킨 TiO2 혼성 시스템도 활발히 연구가 진행되고 있다.
무기 광촉매를 광전극으로 활용하여 광에너지와 전기에너지를 동시에 활용하는 광전기화학 시스템은 InP, GaP, GaAs, CdTe 등의 p형 광촉매들을 중심으로 연구가 진행되고 있다. 광전기화학시스템의 경우 광화학 시스템에 비해 이산화탄소 전환효율이 상대적으로 매우 높은 장점을 가지고 있으나, 광촉매 물질을 그대로 광전극으로 사용할 경우 광전기화학 반 응임에도 불구하고 일반 전기화학적 반응과 마찬가지로 매우 높은 개시전위가 요구된다는 단점이 있다. 그러나 광전극 표면에 여러 가지 금속 나노입자를 도핑하거나 균일계 분자촉 매를 결합시킬 경우 이산화탄소 환원반응에 요구되는 개시전 위를 현저히 낮추거나 혹은 생성되는 환원 부산물의 조성을 다양하게 변화시킬 수 있는 것으로 나타나, 앞으로 다양한 촉매와 광전극의 조합을 통해 보다 효율적인 이산화탄소 전환 시스템을 구축할 수 있을 것으로 기대된다.
이산화탄소 전환 기술은 지속 성장을 위한 온난환 문제와 자원 고갈을 대비해서 시급히 개발되어야 한다. 앞으로 상용화 가능성이 가장 큰 기상 전환 기술은 보다 저에너지 등 온 화한 조건에서 이뤄질 수 있어야 하며 기상 생성물을 분리 정제할 수 있는 저에너지 분리 기술의 개발이 요구되고 있다. 태양 에너지를 직간접적으로 이용하는 광화학적 액상 전환의 경우, 보다 전환 속도 및 효율을 높일 수 있는 방안에 대한 지속 적인 연구가 필요하다. 현재 많은 연구에도 불구하고 이들 전환기술이 아직 경쟁력을 갖추기에는 다소 부족하지만 온실가스 전환 기술은 온실가스 문제 해결 또는 완환 그리고 탄소 자원 확보 측면에서 매우 매력적인 기술임을 알 수 있다. 또한 전환 기술은 신재생 에너지와 바이오메스 그리고 자원 순환 기술의 연계 기술로 그 효과가 더욱 클 것이다.

![Scope of chemical processes for CO2 conversion[1].](http://oak.go.kr/repository/journal/11683/CJGSB2_2012_v18n3_229_f001.jpg)
![Use of carbon dioxide in the chemical industry for the synthesis of organic compounds[2]](http://oak.go.kr/repository/journal/11683/CJGSB2_2012_v18n3_229_t001.jpg)
![Comparison of catalytic activity for carbon dioxide dry reforming of methane[3]](http://oak.go.kr/repository/journal/11683/CJGSB2_2012_v18n3_229_t002.jpg)
![[2+2] Cycloaddition, metal-imdo complex synthesis.](http://oak.go.kr/repository/journal/11683/CJGSB2_2012_v18n3_229_f004.jpg)
![AE factor for various DMC routes[36]](http://oak.go.kr/repository/journal/11683/CJGSB2_2012_v18n3_229_t003.jpg)
![Fraction of products vs. dielectric constant obtained by irradiation of (A) Q-TiO2/SiO2 for 3 h and (B) P-25 TiO2 for 18 h. The solution contained 1 M 2-propanol in all cases except for the use of 2-propanol as the solvent. Solvents used were: (a) carbon tetrachloride, (b) dichloromethane, (c) 2-propanol, (d) ethylene glycol monoethyl ether, (e) acetonitrile, (f) sulfolane, (g) propylene carbonate, (h) water[72].](http://oak.go.kr/repository/journal/11683/CJGSB2_2012_v18n3_229_f010.jpg)
![Correlation between the yield of methanol and the conduction bands of semiconductor catalyst[63].](http://oak.go.kr/repository/journal/11683/CJGSB2_2012_v18n3_229_f011.jpg)