Polyimide (PI) is considered as an important high-performance polymer owing to its excellent properties, such as its high mechanical properties, superior thermal stability, good chemical resistance, low dielectric constant, and good processability [1,2]. Therefore, it has been widely used in the automotive, microelectronic, optic, and membrane fields [3]. However, the accumulation of electrostatic charge as well as its poor heat dissipation must be resolved in certain PI applications, and these issues have been addressed by dispersing nanomaterials such as silica nanoparticles, metal nanoparticles, and especially carbon nanotubes (CNTs) into the PI matrix [4-7]. Although CNTs are effective fillers to enhance the mechanical and electrical properties of PI, they cannot be dispersed easily in a solvent or a polymer matrix due to the van der Waals force [8,9].
Graphene, a two-dimensional (2D) carbonaceous material with sp2-bonded carbon atoms, has attracted much attention since its experimental discovery in 2004 due to both its high aspect ratio and high surface area [10-12]. For composite applications, the significant van der Waals force of pure graphenes such as CNTs presents an obstacle when attempting to disperse them in both organic and inorganic solvents [13]. In contrast, graphene oxide (GO) obtained from graphite by chemical oxidation can meet this demand. It is well dispersed in organic solvents due to the functional groups on its basal planes and edges, such as epoxides, hydroxyls, ketones, diols and carbonyl groups [12,14-17]. In addition, GO is a highly cost-effective material compared to graphene or CNTs because it can be obtained from graphite, which is still abundant in nature after a simple preparation treatment.
GO has been chemically modified to improve degree of dispersion in a solvent, and a small amount of GO has been incorporated into a polymer matrix by a solution-casting method [18-20]. Luong et al. [18] reported the mechanical and electrical properties of PI composites containing 0.38 or 0.75 wt% of ethyl-isocyanate-treated GO (iGO). A significant increase in the electrical conductivity (8.9 × 10-5 S/m) was observed with only 0.75 wt% of iGO. Kim et al. [19] modified GO using γ-aminobutyric acid to introduce carboxylic acid groups, and GO up to 0.8 wt% was should to be well dispersed into PI. However, the composite showed a significant decrease in its tensile strength compared to pure PI (from 117.8 ± 5.4 to 81.3 ± 4.3 MPa). Other polymers such as poly(vinyl alcohol) have been used for GO composites to improve their mechanical and barrier properties, but an increase in the electrical conductivity was not reported [20].
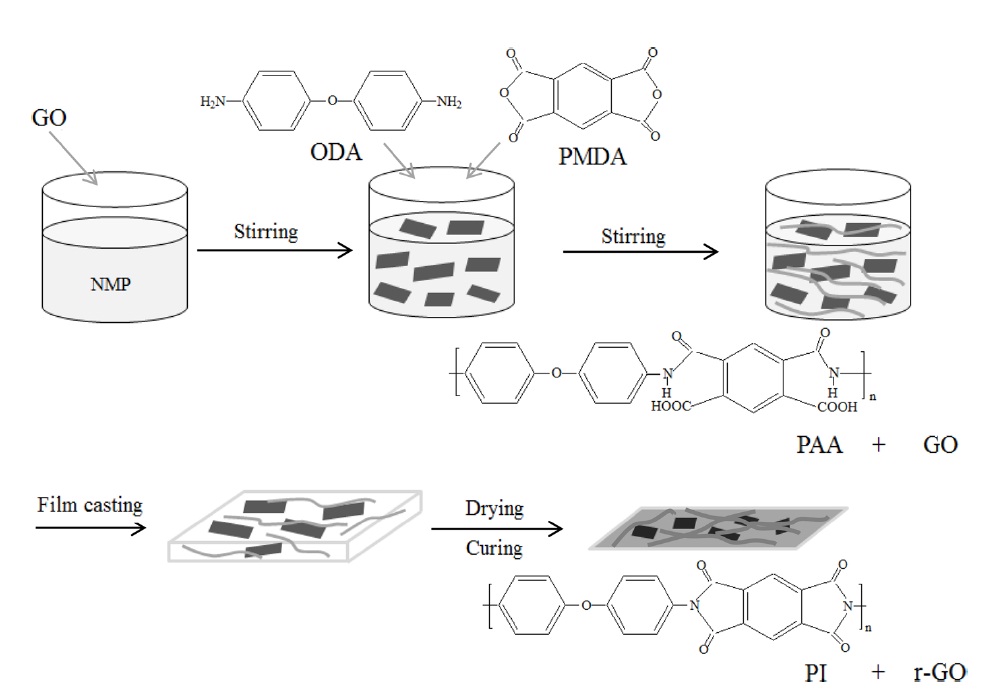
In this study, composites containing a high content of GO were prepared to observed their mechanical and electrical properties. Reduced GO/PI composite films containing 10 or 20 wt% of GO, respectively, were prepared via a conventional solution-casting method. GO was converted into a reduced form of GO (r-GO) during the imidization process. The synthesis of the polymer was confirmed by Fourier transform-infrared spectroscopy. The heattreated GO underwent a thermogravimetric analysis in order to mimic the imidization and to investigate the reduction of GO. The volume resistivity and tensile properties were measured by a four-point probe unit and a universal tester, respectively. After the tensile test, the fractured surfaces ofthe composite films were observed by a scanning electron microscope.
Graphite flakes (325 mesh, 99.8%) were obtained from Johnson Matthey Co. 4,4'-oxydianiline (ODA, 97%), pyromellitic dianhydride (PMDA, 97%), N-Methyl-2-pyrrolidone (NMP, 99.5%) and potassium permanganate (KMnO4, 99.0%) was purchased from Sigma-Aldrich Co. Sulfuric acid (H2SO4, 97%) and hydrogen peroxide (H2O2, 30 wt%) were purchased from Matsunoen Chemicals LTD and from Daejung Chemical & Metals Co., LTD, respectively. All of the above chemicals were used as received.
GO was synthesized by a modified Hummers method [21]. Four grams graphite flakes were added to a 250 mL round-bottom flask containing 120 mL H2SO4 and this was stirred for 1 h. Fifteen grams KMnO4 was divided into 9 parts which were then added to the mixture at intervals of 20 min while being stirred. The mixture was slowly heated to 40℃ and maintained for 5 h in order to oxidize the graphite. Subsequently, 150 mL of deionized (DI) water was added to the mixture. A 15 mL H2O2 solution was added to the mixture under stirring for 30 min and then kept in this condition for 24 h. Next, the mixture was centrifuged. After that, the mixture deposed in the dialysis tube was washed with DI water several times to obtain a pH of 5. Finally, the GO was dried in a freeze dryer at -45℃ for 24 h.
2.2. Synthesis of poly(amic acid) and its composite solutions
The precursor of PI, poly(amic acid) (PAA), was synthesized from 0.920 g PMDA and 0.845 g ODA at an equivalent molar ratio. The poly-condensation step was performed in 10 g of NMP at room temperature under magnetic stirring for 24 h. For composite solutions, 0.177 g and 0.353 g of the obtained GO were dispersed in 10 g of NMP by ultra-sonication for 4 h to prepare 10 wt% (compared to all solids) and 20 wt% GO/PAA composites, respectively. Next, equivalent molar ratios of 0.760 g ODA and 0.828 g PMDA were dissolved in a GO dispersion (containing 0.177 g GO), and 0.676 ODA as well as 0.736 g PMDA were dissolved in a GO dispersion (containing 0.353 g GO). The poly-condensation process was identical to that of the pure PAA.
2.3. Preparation of pure PI and its composites
Pure PI and r-GO/PI composite films were prepared by conventional solvent casting, after which the imidization of the PAA and GO/PAA composite films was carried out. The PAA and GO/PAA solutions were cast onto commercial PI films with a thickness of ~800 μm. All of the cast films were initially dried at 90℃ for 3 h in a vacuum oven to remove the residual solvent, after which they were thermally imidized using the following procedure: (1) heating to 100℃ for 33 min and maintaining at 100℃ for 1 h, (2) heating to 200℃ for 33 min and maintaining at 200℃ for 1 h, (3) heating up to 300℃ for 33 min and maintaining at 300℃ for 1 h, and then cooling naturally to room temperature. The film thickness was found to be ~30 μm.
Fourier transform-infrared spectroscopy (FT-IR Spectrophotometer, Nicolet IS10, USA) was performed to confirm the synthesis of PAA and PI. The volume resistivity was measured using a resistivity meter (FPP-RS8, DASOL ENG, Korea) and a super megohmmeter (SM-8200, Hioki, Japan). A thermogravimetric analysis (TGA) was conducted to investigate the reduction of GO in a nitrogen atmosphere at a heating rate of 10℃ /min using a TGA Q50 device (TA Instruments, USA). The tensile testing of the films was carried out using an Instron Universal Tester 5567 (Instron, USA) with a cross head speed of 5 mm/min. The length and width of the specimen were 2.5 cm and 0.5 cm, respectively, and at least 10 specimens were prepared for each test. Field-emission scanning electron microscopy (FE-SEM, Hitachi S-4700) performed at an acceleration voltage of 15 kV was used to observe the morphology of the fractured surfaces regarding pure PI and its composites after tensile testing.
GO was prepared by the modified Hummers method and then dispersed in NMP solvent by ultrasonication for a uniform dispersion. The GO dispersion was used as the filler in the synthesis of PAA. Next, the GO/PAA solutions were cast onto commercial PI film in conjunction with a heat treatment to obtain r-GO/PI composite films. Scheme 1 demonstrates the steps to fabricate these films. A nucleophilic attack of the amino group in ODA on
the carboxyl carbon of the anhydride group in the PMDA led to the opening of the anhydride ring, forming an amic acid group [22]. Water molecules were then removed during the imidization process through the heat treatment, and PI and its composite films were produced.
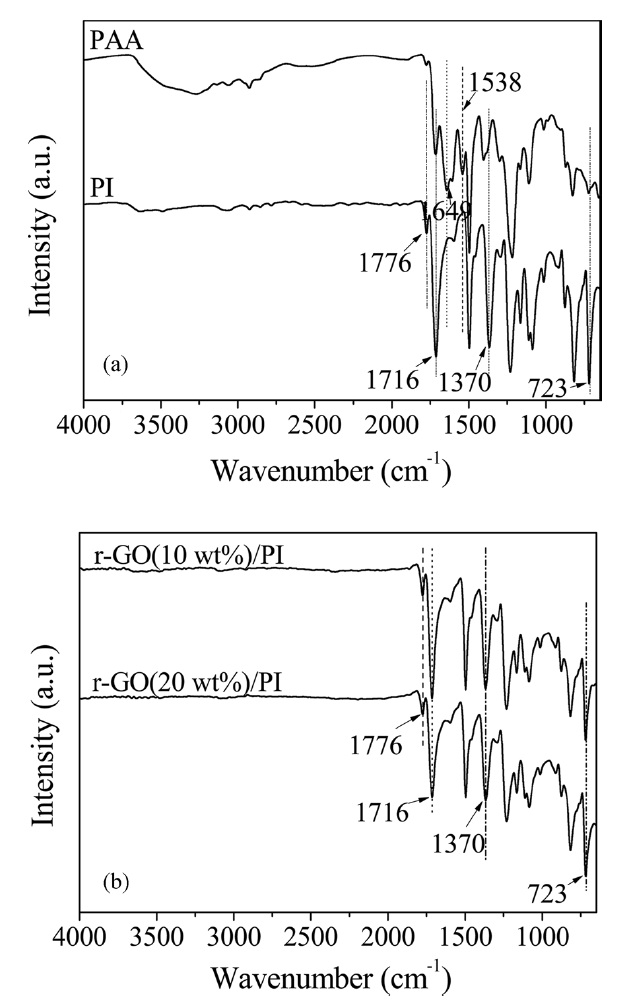
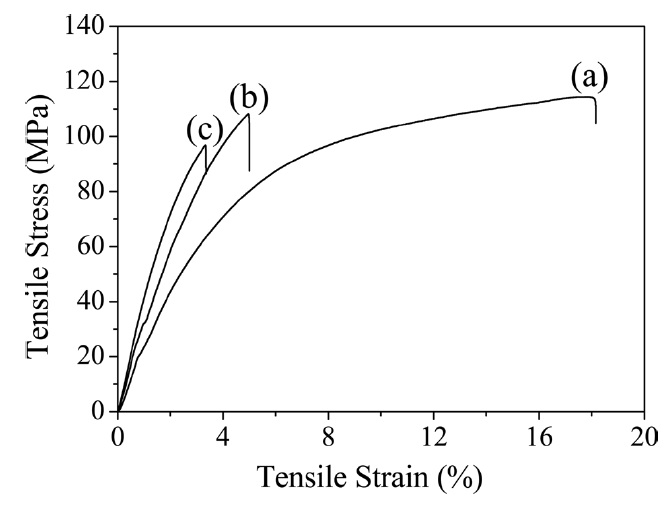
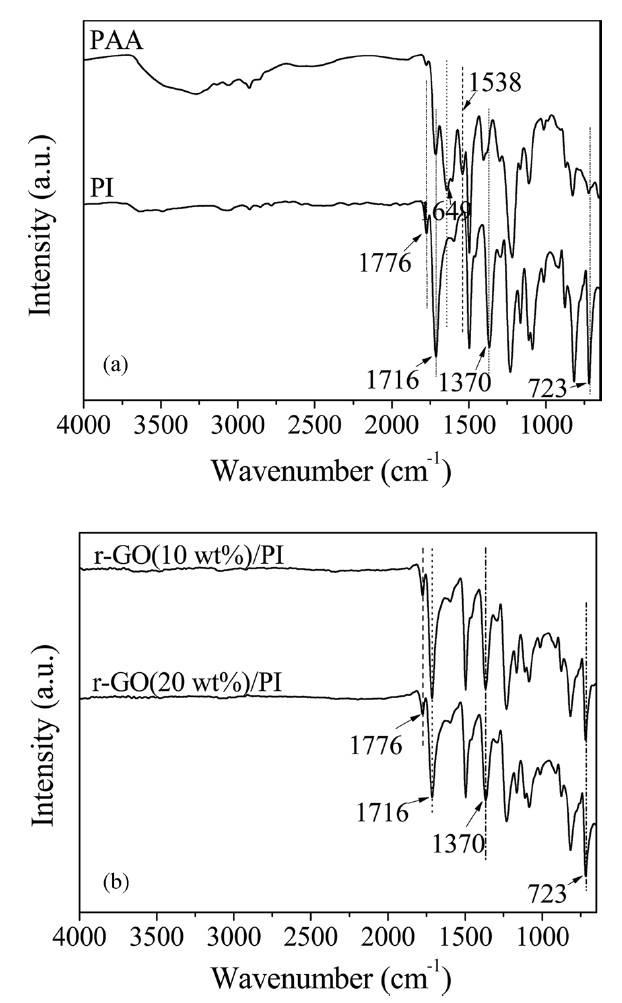
The FT-IR spectra of PAA, PI and its composites are shown in Fig. 1. Prior to heating, the absorption peaks at 1649 and 1538cm-1 were assigned to the C-NH stretching (amide II) and C=O in the CONH (amide I) stretching bands [6]. In the FT-IR spectrum of PI, the absorption peaks at 1650 and 1539 cm-1 disappeared and significant absorption bands at 1776 cm-1 (C=O asymmetric stretching of imide), 1716 cm-1 (C=O symmetric stretching), 1370 cm-1 (C-N stretching) and 723 cm-1 (C=O bending of imide) were observed, indicating the imide group and the completion of imidization [7,23,24]. The FT-IR spectra of the r-GO/PI composites also showed the characteristic absorptions of the imide unit at 1776, 1716, 1370 and 723 cm-1. However, there was no significant difference between the PI and its composites. These results confirmed that r-GO/PI composite films were successfully obtained.
3.2. Volume resistivities of pure PI and r-GO/ PI composite films
The volume resistivities of pure PI and r-GO/PI composite films were measured by a four- point probe unit (Table 1). The volume resistivity of pure PI was found to be 3.4 × 1013 Ω?m. However, the volume resistivities were 4.0 × 108 Ω?m and 1.0 × 104 Ω?m for the r-GO/PI composites containing 10 and 20 wt% GO, respectively. The volume resistivity decreased as the concentration of GO increased. Compared to pure PI film, the volume resistivity of r-GO/PI composite film containing 20 wt% of GO oxide decreased by more than nine orders of magnitude due to the electrical percolation networks of the partially reduced GO in the films.
[Table 1.] Electrical and mechanical properties of pure PI and its composites

Electrical and mechanical properties of pure PI and its composites
[Fig. 2.] Thermogravimetric analysis thermograms of (a) graphene oxide (GO) and (b) heat-treated GO.
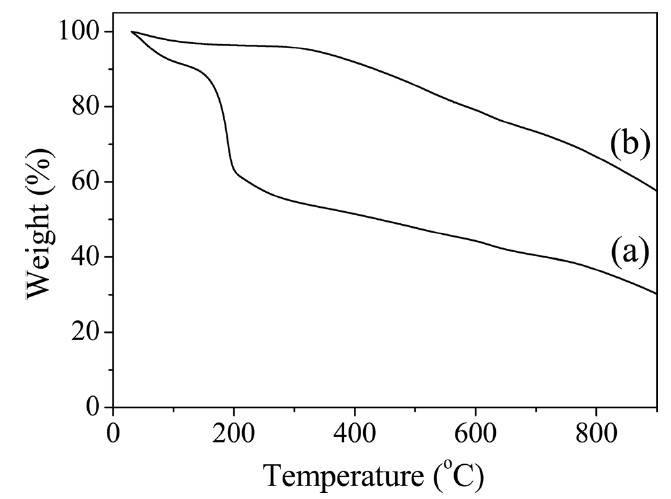
To confirm the reduction of GO as obtained during the imidization process, a TGA was conducted on both the untreated and heat-treated GO prepared under the same imidization condition used with the composites. As shown in Fig. 2, the weight loss of the untreated GO which occurred at the beginning was due to the evaporation of the stored water [25-28]. The majority oft loss, which occurred at approximately 180℃ , was due to the pyrolysis of the labile oxygen-containing functional groups [29]. However, the majority oft loss of the heat-treated GO began when the temperature exceeded 300℃ , which was much higher than the case of the untreated GO, indicating that the GO was partially reduced during the heat treatment. It was evident that the r-GO acted as a conducting filler material in the PI film.
A color change was observed due to the reduction from GO
to r-GO. Figs. 3a and b show photographs of untreated GO and heat-treated GO, respectively. The color changed from brown for GO to black for r-GO during the heat treatment. In addition, photographs of pure PAA and its composite films with different concentrations of GO before and after imidization are shown in Figs. 4a and b, respectively. It is clear that the transparency decreased with an increase in the content of GO, after which the corresponding colors became darker. Furthermore, the composite film containing 10 wt% of GO was transparent. It became opaque after imidization, indicating that the GO underwent a partial reduction in the composite film upon imidization [19].
3.3. Mechanical properties of pure PI and its composite films
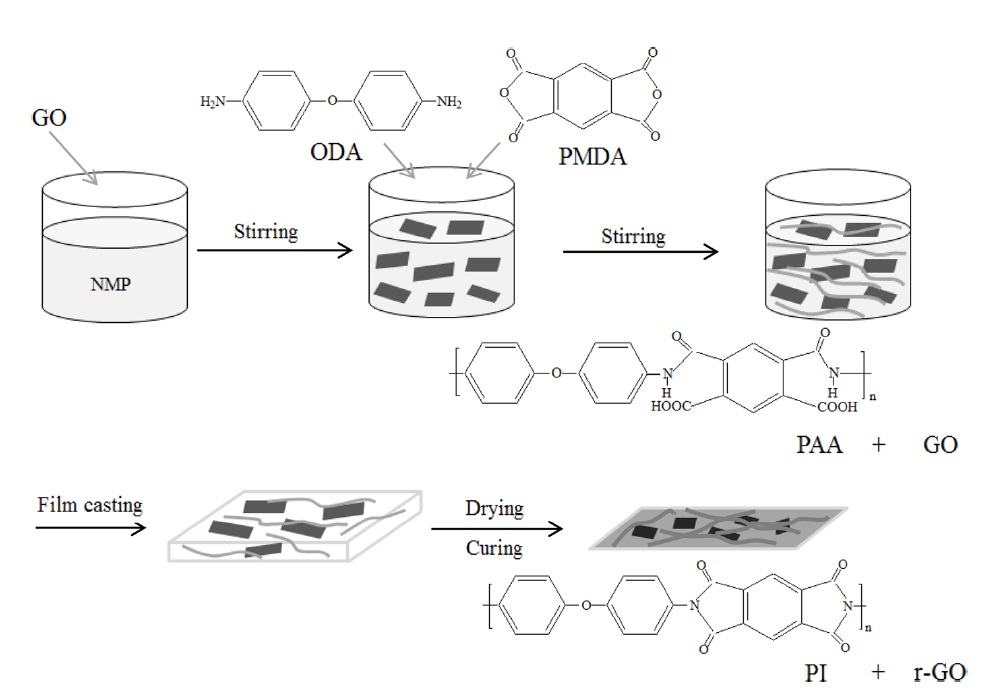
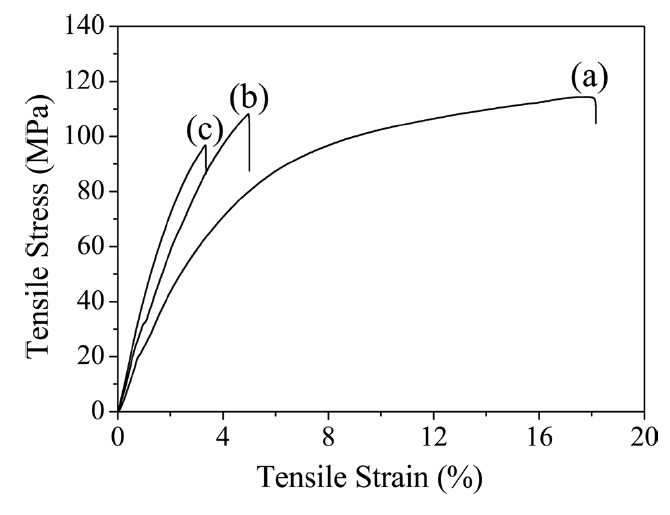
Thermal imidization is an important process to determine the properties of PI, including its mechanical properties [7]. The typical stress-strain curves of pure PI and its composite films are shown in Fig. 5, and the values of the tensile properties are summarized in Table 1. The Young’s modulus increased significantly with the addition of GO. However, the strength and elongation decreased as GO loading increasedg’s modulus with regard to the r-GO/PI composite film containing 20 wt% GO drastically increased from 2.3 GPa to 4.4 GPa most likely due to the non-covalent and molecular level interaction between r-GO and PI [18]. Also, the corresponding tensile strength decreased from 113 MPa to 99 MPa owing to the fillers in the composite, which led to the phase separation of r-GO from the PI matrix [30]. It is well known that external stress on a plastic composite is transferred from the continuous phase (polymer matrix) to the discontinuous phase (filler) and that the final properties depend on the extent of the bonding between the two phases [31]. It was interesting to note that while the GO was highly loaded up to 20
wt% in the PI matrix, the tensile strength decreased by only 12% compared to the pure PI. This indicated that the composites underwent non-covalent interaction between the r-GO and PI phases, resulting in good mechanical properties. The fractured surfaces of the pure PI and r-GO/PI composite films were observed by SEM after tensile testing in order to understand the tensile properties of the r-GO/PI composites, In Fig. 6a, the fractured surfaces of the pure PI film are shown to be fairly smooth. Furthermore, for the composite films containing 10 and 20 wt% of GO shown in Figs. 6b and c, respectively, the fractured surfaces were relatively rough compared to that of the pure PI film [19].
GO was prepared from graphite using a modified Hummers method. The GO was used as nanofillers for the preparation of r-GO/PI composites by an in-situ polymerization method. FT-IR was performed in order to confirm the synthesis and imidization regarding PAA and its composites with GO. The volume resistivity of the r-GO/PI composite film containing 20 wt% of GO was found to be 1.0 × 104 Ω?m, which was nine orders of magnitude less than that of pure PI film (3.4 × 1013 Ω?m). The TGA thermogram showed that the decomposition temperature of the GO which underwent a heat treatment identical to imidization was around 300℃, indicating the partial reduction of GO. Therefore, this suggests that partially reduced GO during imidization served as a conducting filler material to form electrical percolation networks in the composites. The tensile test indicated that the Young’s modulus of the r-GO/PI composite film containing 20 wt% GO was drastically increased from 2.3 GPa to 4.4 GPa, which was approximately an 84% improvement compared to that of pure PI film, whereas the corresponding tensile strength decreased by only 12%, from 113 MPa to 99 MPa.