With the fast growth of biological technology, there has been a rapid growth in the need to process biological data. This data has been made available with lower costs via advanced bio-sensor technologies, and as a result, in the next few years, we expect to witness a dramatic increase in the application of genomics in our everyday lives, such as in our personalized medicine.
Bioinformatics research is interdisciplinary in nature, in that it integrates diverse areas such as biology and biochemistry,data mining and machine learning, database and information retrieval, computer science theory, as well as many others. Bioinformatics concerns a wide spectrum of biological research,including genomics and proteomics, evolutionary and systems biology, etc. These research areas are increasingly dealing with data collected from a wide spectrum of devices such as microarrays,genomic sequencing, medical imaging and so on [1].Based on this data, data mining in bioinformatics targets the investigation of learning statistical models to infer biological properties from new data. Various techniques such as supervised learning and unsupervised learning have been developed,with promising results and biological insights in areas such as sequence classification, gene expression data analysis and biological network reconstruction, etc.
A typical assumption in biological data mining is that a sufficient amount of annotated training data is required, so that a robust classifier model can be trained. In many real-world biological scenarios, however, labeled training data is lacking.Sometimes this data can only be obtained by paying a huge cost. This problem of a lack of labeled training data, which partly causes the so-called data sparsity problem, has become a major bottleneck in practice. When data sparsity occurs, over fitting is common when we train a model. As a result, the statistical model will experience a reduction in performance.
In response to the above problem of data sparsity, various novel machine-learning methods have been developed. Among them is the transfer learning framework, which refers to a new machine learning framework which reuses knowledge from other domains. Transfer learning aims to extract knowledge from some auxiliary domains where the data is annotated, but it cannot be directly used as the training data for a new domain.To reuse this data, transfer learning compares auxiliary and target problem domains in order to find useful knowledge that is common between them, and then reuses this knowledge to help boost learning performance in the target domain. A subarea of transfer learning is multi-task learning, where the target task and the source task are treated the same, and are learned simultaneously.Multitask learning stresses generating benefits for learning performance improvements in all related domains.Transfer learning and multi-task learning have achieved great success in many different application areas over the last two decades [2, 3], including text mining [4-6], speech recognition[7, 8], computer vision (e.g., image [9] and video [10] analysis),and ubiquitous computing [11, 12].
In this article, we give a survey on some recent transfer learning methods in bioinformatics research, together with an additional overview on biomedical imaging and sensor based ehealth.We will cover some of the most important application areas of bioinformatics, including sequence analysis, gene expression data analysis, genetic analysis, systems biology and biomedical applications in bioinformatics. By conducting a survey in these areas, we hope to take stock of the progress made so far, and help readers navigate through technological advances in the future.
II. DATA MINING IN BIOINFORMATICS: A SURVEY OF PROBLEMS
We focus on the following biological problems in this survey:sequence analysis, gene expression data analysis and genetic analysis, systems biology, biomedical applications.
>
A. Biological Sequence Analysis
Biological sequence analysis aims to assign functional annotations to sequences of DNA segments, and is important in our understanding of a genome. One example is the identification of splice sites in terms of the exon and intron boundaries, a complex task due to the number of alternative splicing possible.Other examples include the prediction of regulatory regions that allow the binding of proteins and determine their functions; the prediction of transcription start and initiation sites, and the prediction of coding regions. Another important sequence analysis problem is the major histocompatibility complex (MHC) binding prediction from a sequence perspective, where sequences from pathogens provide a huge amount of potential vaccine candidates [13]. MHC molecules are key players in the human immune system, and the prediction of their binding peptides helps in the design of peptide-based vaccines, but there is a major lack of quality labeled training data that can be used to build a good prediction model for this binding prediction problems.In addition, protein subcellular localization prediction problems are also important in biological sequence analysis, but annotated locations are difficult to obtain.
>
B. Gene Expression Analysis and Genetic Analysis
Gene expression analysis and genetic analysis through microarrays or gene chips is an important task for the understanding of proteins and mRNAs. A microarray experiment measures the relative mRNA levels of genes, which allows us to compare the gene expression levels of some biological samples over time in order to understand the differences between normal cells and cancer cells [14]. One characteristic of this analysis is that the number of features that correspond to genes is usually more than the number of samples. This makes it difficult to apply traditional feature selection approaches directly to this data to reduce its dimensionality [15]. Another data mining task often applied to gene expression data is co-clustering, which aims to cluster both the samples and genes at the same time[16]. In genetic analysis, recent advances have allowed genome-wide association studies (GWAS) that can assay hundreds of thousands of single nucleotide polymorphisms (SNPs)and relate them to clinical conditions or measurable traits. A combination of statistical and machine learning methods has been exploited in this area [17, 18].
>
C. Computational Systems Biology
Computational systems biology refers to the tasks of modeling gene-protein regulatory networks, and making inferences based on protein-protein interaction networks. The computational challenge relates to the task of integrating and exploring large-scale, multi- dimensional data with multiple types. An important issue is how to exploit the topological features of the network data to help build a high-quality model. Besides automatic prediction based on statistical models, mixed initiative approaches such as visualization have also been developed to tackle the large-scale data and complex modeling problems.
Biomedical applications investigated in this survey include biological text mining, biomedical image classification and ubiquitous healthcare. Biological text mining refers to the task of using information retrieval techniques to extract information on genes, proteins and their functional relationships from scientific literature [19]. Today we face a vast amount of biological information and findings that are published as articles, journals,blogs, books and conference proceedings. PubMed and MEDLINE provide some of the most up to date information for biological researchers. A scientist often has to read through a large volume of published text in order to understand and find any potential knowledge embedded in the text. With text mining technology, many new findings that are published in text format,such as new genes and their relationships, can be automatically detected, which helps man and machine work together as a team.
Furthermore, biomedical image classification is an important problem that appears in many medical applications. Manual classification of images is time-consuming, repetitive, and unreliable.Given a set of training images labeled into a finite number of classes, our goal is to design an automatic image classification method to understand future images. An example of this is breast-cancer identification from medical imaging data using computer-aided detection on the screening mammography.An important issue for such models is how to reduce the false-positive rates in the classifications.
Ubiquitous computing is increasingly influencing health care and medicine and aims to improve traditional health care [20]. Exercises are essential to keep people healthy. Recently, several devices have been designed to record and monitor users’ routine exercises. For example, the accelerometer [21-23] and gyroscope[24-26] are used to recognize the users’ activities; Polar records heart rate to evaluate the amount of activity in terms of calories in order to manage sports training [27]. Pedometers can quantify activities in terms of number of steps, and then suggest the users to reduce or increase the amount of exercise they undertake [28].
In all the above-mentioned problems, transfer learning plays an important role for solving the data sparsity problem. Below,we survey some recent transfer learning studies in these areas.To start with, we first introduce some notations first that are used in this survey. The source domain data
III. BIOLOGICAL SEQUENCE ANALYSIS
In sequence classification, the goal is to annotate gene sequences or protein sequences from a given set of training data. As we mentioned above, the process of learning to annotate sequences often suffers from a lack of labeled data, which often leads too verfitting. To solve this problem, multi-task learning methods are often used, where it possible to learn to annotate two or more sets of sequence data together. In this approach, the sequence data can be from different problem domains. By learning these tasks together, the lack of labeled data problem is alleviated.
In multi-task learning, task regularization is an often used.Task regularization methods are formulated under a regularization framework, which consist of two objective function terms:an empirical loss term on the training data of all tasks, and a regularization term that encodes the relationships between tasks. We first introduce a general framework of regularization based multi-task approach. In their pioneering work, Evgeniou and Pontil [29] proposed a multi-task extension of support vector machines (SVMs), which minimizes the following objective function,

The first and second terms of Eq. (1) denote the empirical error and 2-norm of parameter vectors, respectively, which are the same as those in single-task SVMs. In biological sequence classification tasks, these terms refer to the errors committed when making a comparison to the training data. The difference between single-task SVMs and multi-task SVMs lies in the third term of Eq. (1), which is designed to penalize large deviations between each parameter vector and the mean parameter vector of all tasks. The penalized term imposes a constraint that the parameter vectors in all tasks be similar to each other. In multi-task sequence classification problems, this term characterizes the extent of knowledge sharing between the learning tasks in different sequences. With this general background on SVM-based multitask learning in mind, we now sample a few well-known works using regularization based approaches, as well as kernel design for biological sequence analysis. We will pay attention to applications in splice site recognition and MHC binding prediction.
One of the first works in multi-task biological sequence classification was by Widmer et al. [30], who proposed two regularization based multi-task learning methods to predict the splice sites across different organisms. In order to leverage information from related organisms, Widmer et al. [30] suggested two principled approaches to incorporate relations across various organisms. The proposed methods were implemented by modifying the regularization term in [29]. However, the relations between the organisms in [30] was defined by a tree or graph implied by their taxonomy or phylogeny. The models related to task t served as prior information for the model of t, such that the parameter
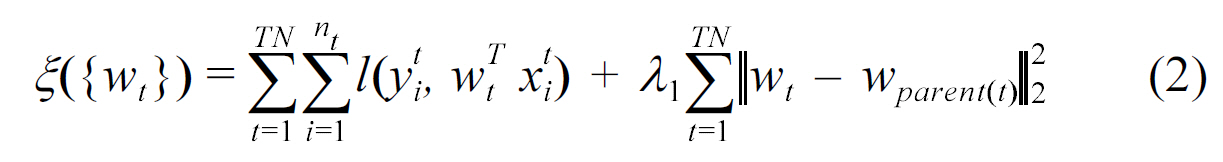
In biology, an organism and its ancestors should be similar due to inheritance of properties in evolution. This is imposed as a constraint in the above formula. Another approach adopted by Widmer et al. [30], is the constraint that ∥ wt ? wt ┃ must be small for related tasks t and t' in their formulation.
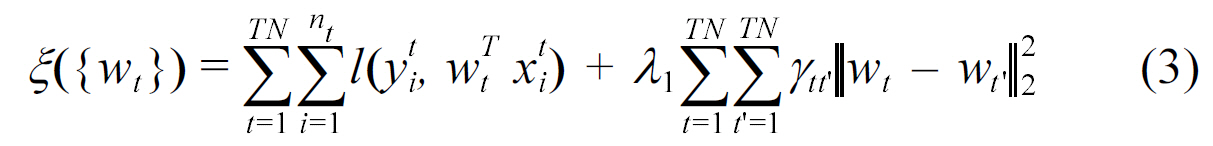
Intuitively, the above formulation states that the properties associated with similar entities are close to each other.
In addition to the above formulations, Widmer et al. [30] also designed a kernel function that not only considers the data of the task

In Eq. (4),
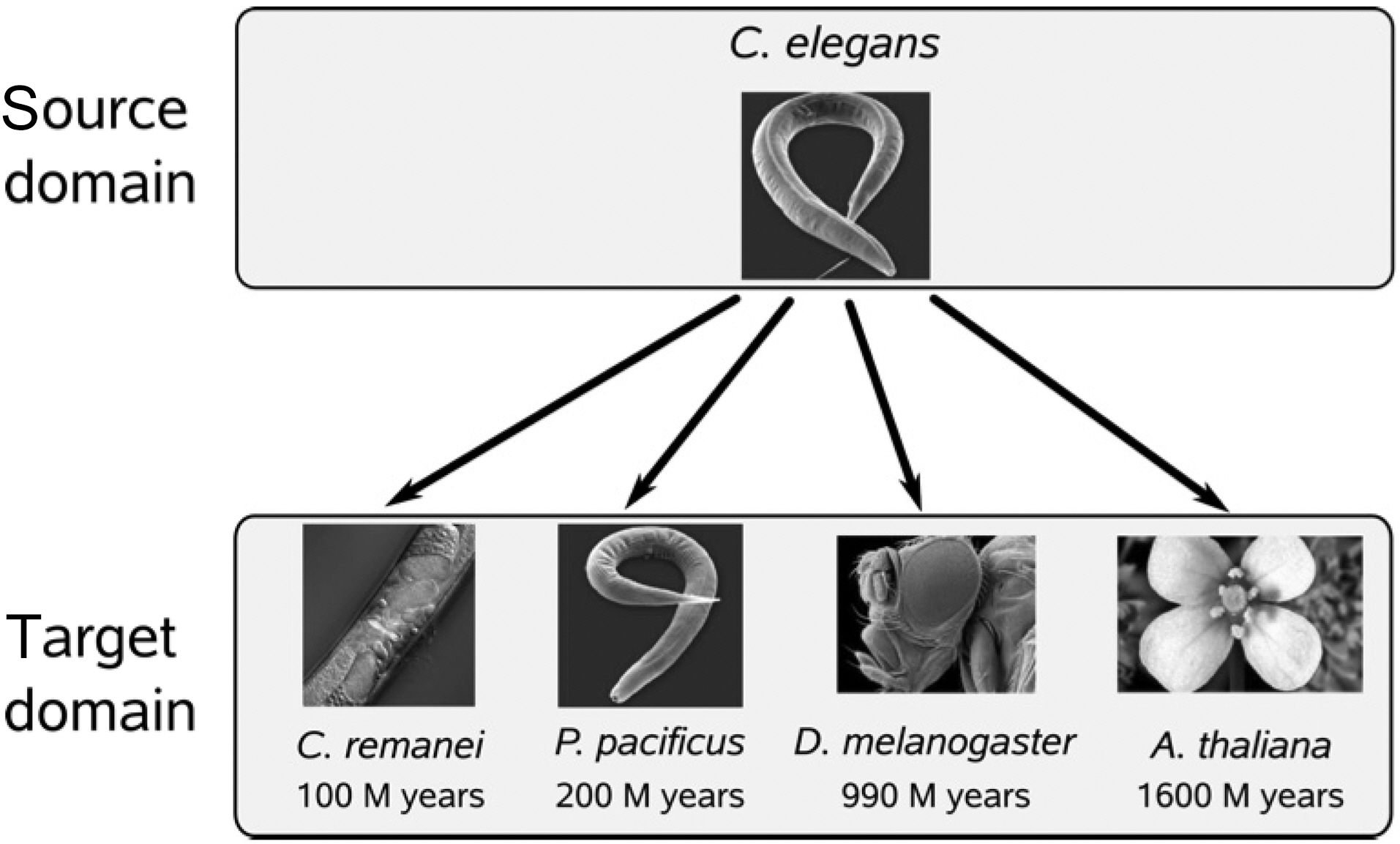
Schweikert et al. [31] considered a number of domain transfer learning methods for splice-site recognition across several organisms. The goal of domain transfer learning is to use a model from a well-analyzed source domain and its associated data to help train a model in the target domain. In the formulation of Schweikert et al. [31],
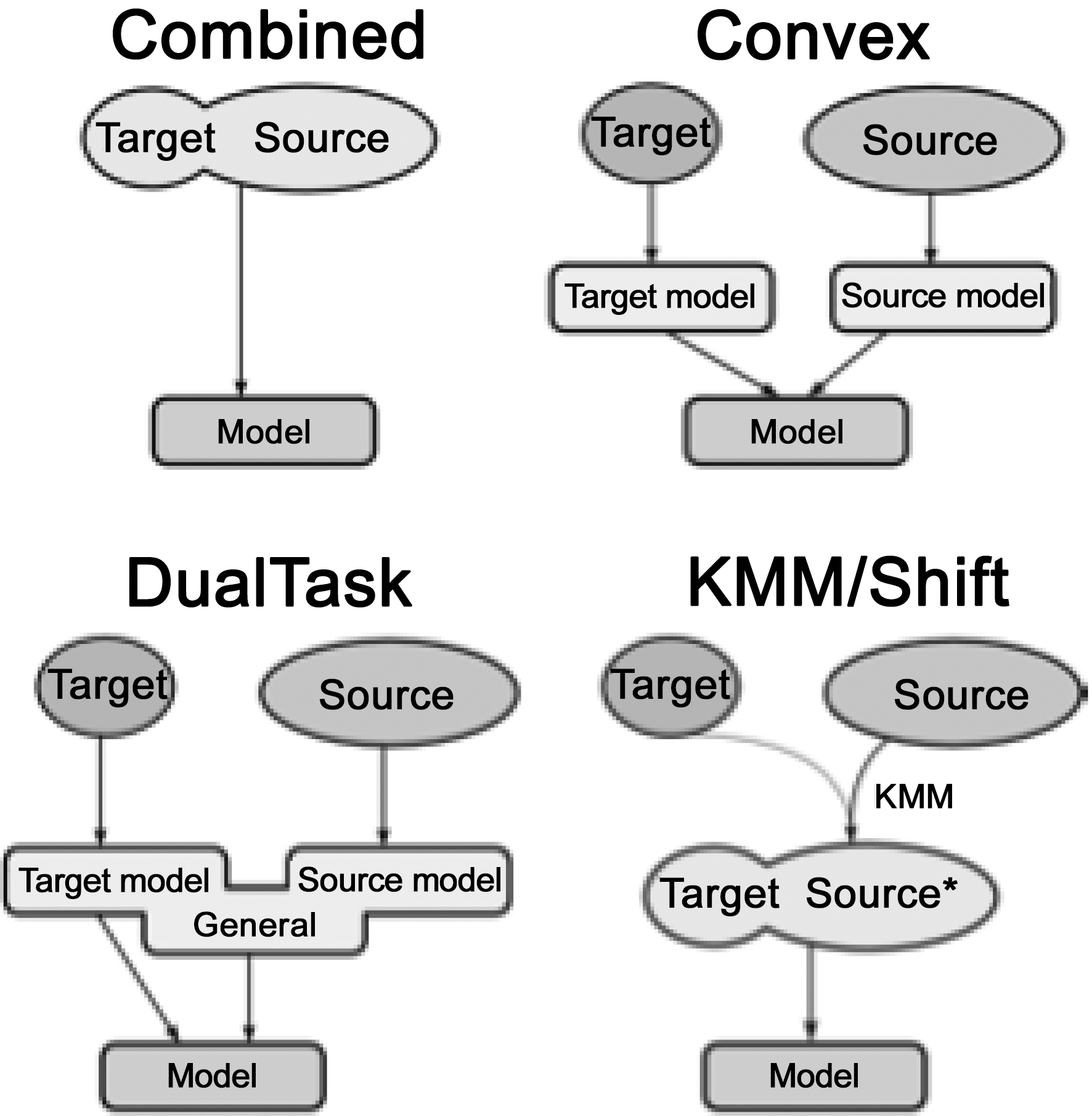
The domain transfer learning methods include (as illustrated in Fig. 2):
- Combination: As a baseline, the simplest way is to combine the source domain data and the targetdomain data directly without considering the weights.
- Convex combination:
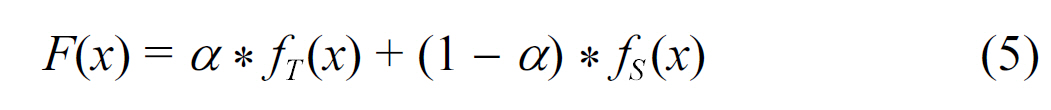
where
- Dual-task learning:
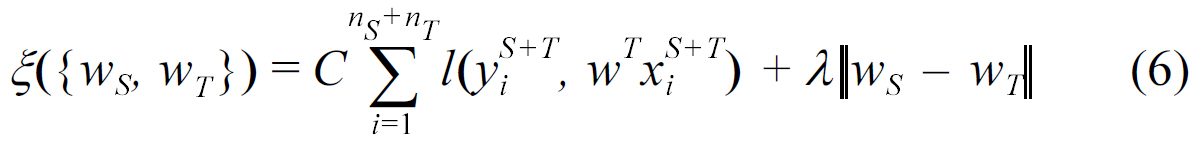
where both of
- Kernel mean matching:
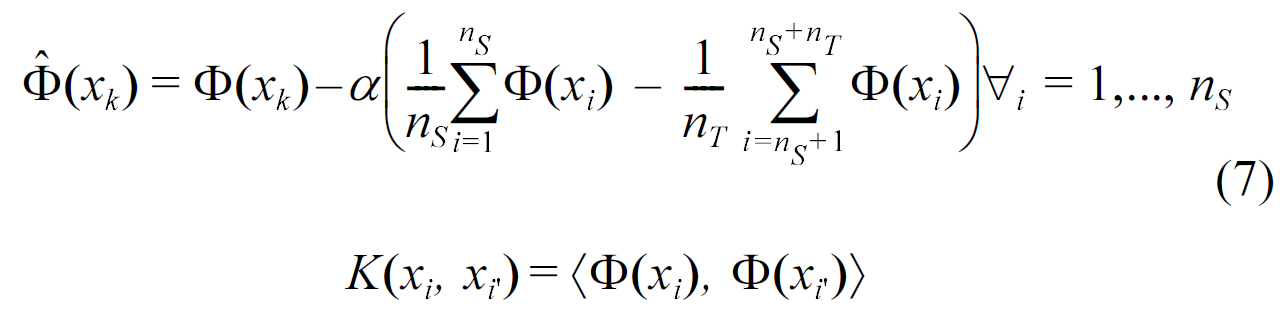
where Φ is the kernel mapping, which projects the data into a reproducing kernel Hilbert space.
Experimental results presented in [31] showed that the differences of classification functions for recognizing splice site in these organisms will increase with increasing evolutionary distance.
Jacob and Vert [32] designed an SVM algorithm that is able to learn peptide-MHC-I binding models for many alleles simultaneously,by sharing binding information across alleles. The sharing of information is controlled by a user-defined measure of similarity between alleles, where the similarity can be defined in terms of supertypes, or more directly by comparing key residues known to play a role in the peptide-MHC binding. Jacob and Vert first represented each pair of alleles
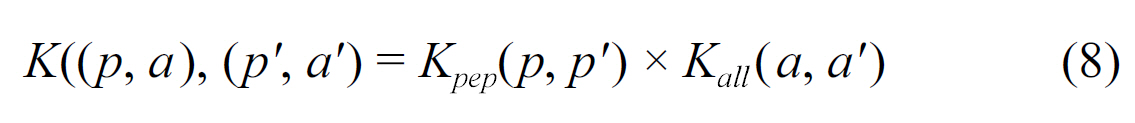
where for the peptide kernel
In the work of Jacob et al. [33], regularization based models are used for MHC class-I binding prediction. A novel, multitask learning method was implemented by designing a norm or a penalty term over the set of weights, such that the weight vectors of the tasks within a group are similar to each other. To achieve this goal, the penalty function was formulated as follows:
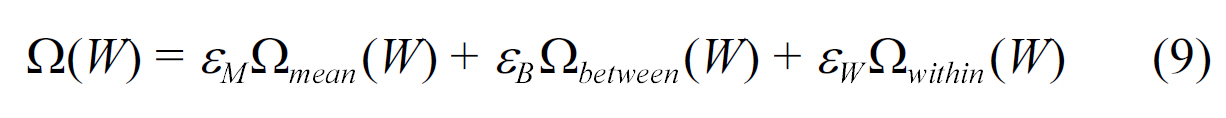
where
Following the pioneering works of Jacob and Vert [32],Jacob et al. [33], and Widmer et al. [34] in 2010 aimed to improve the predictive power of the multitask kernel method for MHC class I binding prediction by developing an advanced kernel based on Jacob and Vert’s orginal. In addition, Widmer et al. [35] investigated multi-task learning scenarios where there exists a latent structure shared across tasks. They modeled the crossover between tasks by defining meta-tasks, so that information is transferred between two tasks t and
As mentioned in Section II, protein subcellular localization prediction can be categorized as biological sequence analysis.Xu et al. [36] compared a multitask learning method with a common feature representation. A common feature representation approach was adapted from Argyriou et al. [37, 38]. In the latter paper, Argyriou et al. [38] proposed a multi-task feature learning method to learn common representations for multi-task learning under a regularization framework:
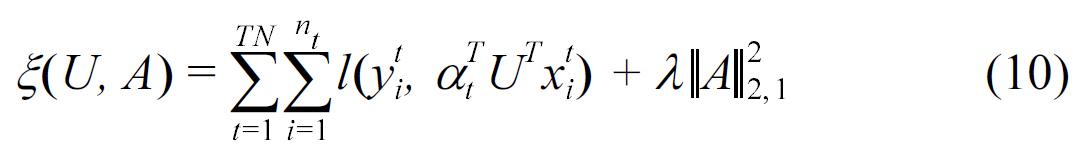
In the above formulation, the first term
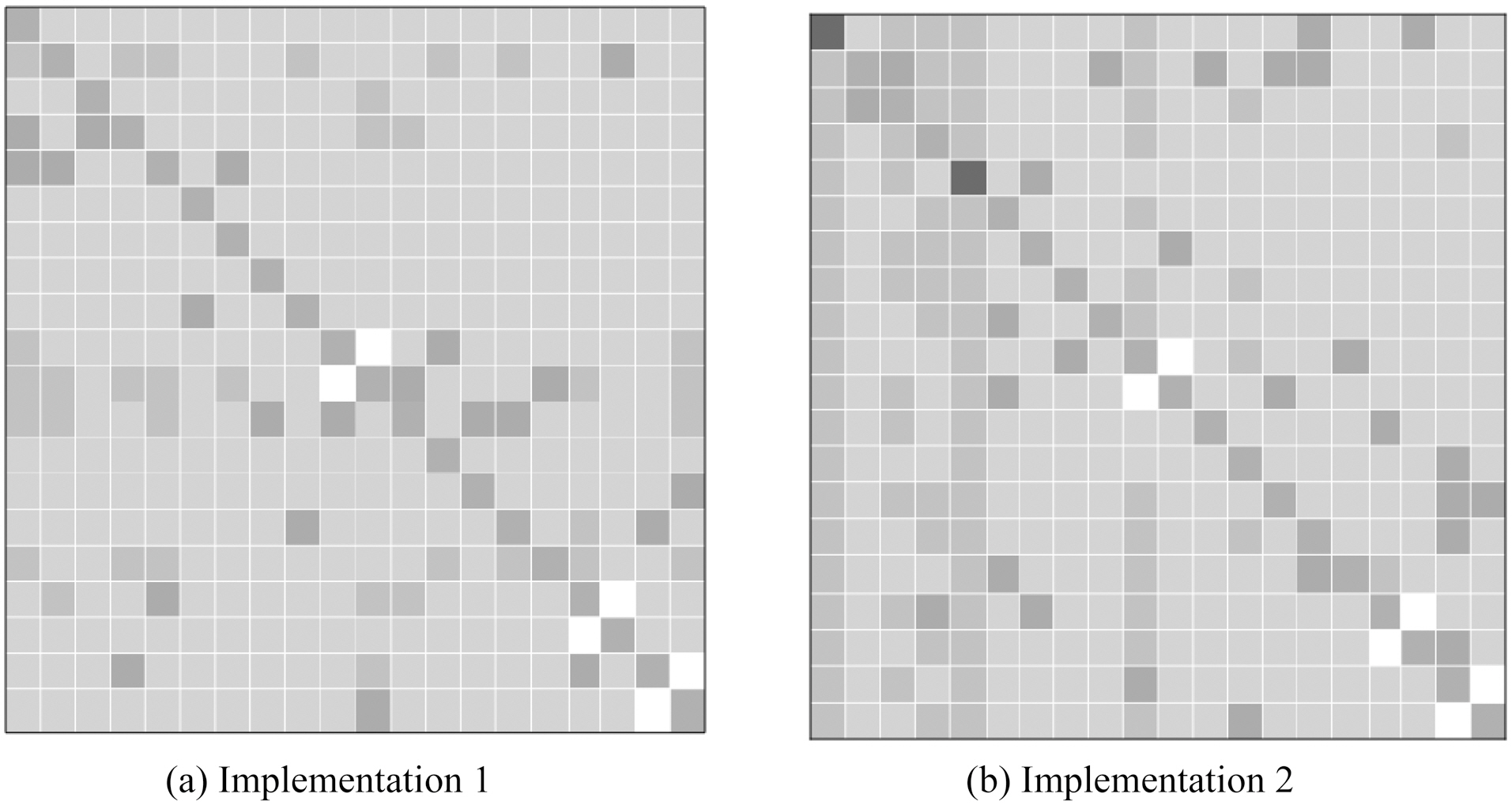
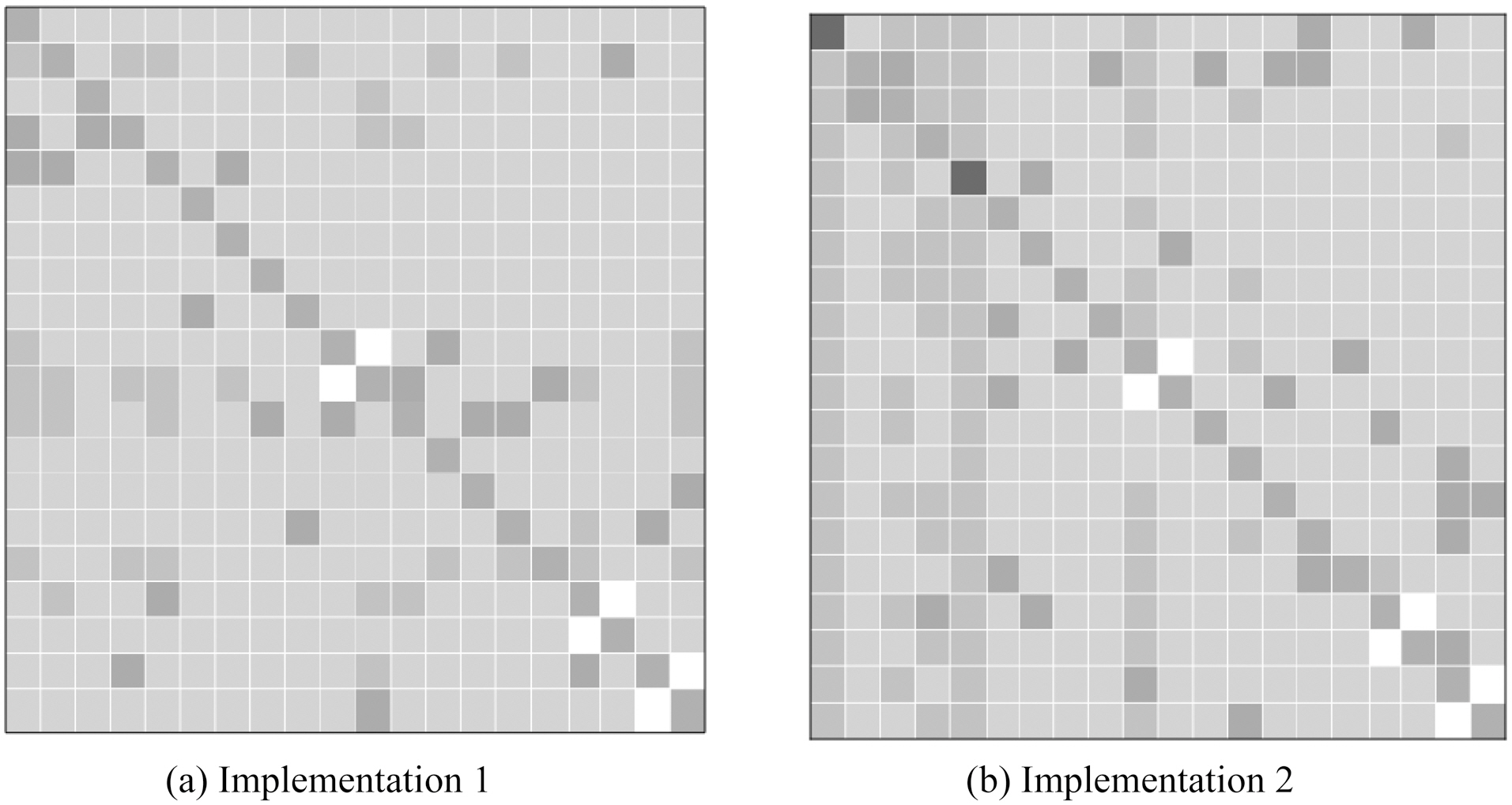
Xu et al. [36] conducted extensive experiments to answer the question: “Can multi-task learning generate more accurate classifiers than single task learning?” They compared the accuracies of the test data between the proposed multi-task learning methods and other prediction model baselines. An example of their results is summarized in Fig. 3, this illustrates the accuracies of multi-task feature learning and those of single task learning.The columns from left to right and rows from up to down represent different organisms: archaea, bacteria, gneg, gpos, bovine,dog, fish, fly, frog, human0, human, mouse, pig, rabbit, rat,fungi, plant0, plant, virus0 and virus in order. Each cell
i as well; these were used as baselines. The cells marked in red indicate that applying multi-task learning methods gives worse performance than the baselines, whereas those in light green indicate that applying multi-task learning methods achieves better performance than the baselines. Furthermore, the cells in dark green or dark grey represent the best performance when evaluating the test data from each organism in a particular column.Finally, the cells in white mean the performance result is missing because some organisms overlap, as in the case of human0 vs. human, plant0 vs. plant and virus0 vs. virus. For these pairs, one cannot conduct multi-task learning experiments.
As shown in above figures, the most significant improvement when using the multi-task learning strategy was about 25%. The performance of plants, viruses and animals can be improved by around 10% by using multi-task learning methods. The columns from left to right and rows from up to down in the table show different organisms: archaea, bacteria, gneg, gpos, bovine, dog,fish, fly, frog, human0, human, mouse, pig, rabbit, rat, fungi,plant0, plant, virus 0 and virus in order. This order arranges learning tasks based on organisms in super type order; for example, those organisms that are animals were put together.Moreover, better results are often obtained near the diagonals,where organisms are similar, while worse cases were often located in the cells far from the diagonals. The results in cells near the diagonals were obtained by training using two relatively similar tasks, like dog and fly, bacteria and archaea, and so on. To conclude, multi-task learning techniques can generally help improve the prediction performance for protein subcellular localization in comparison with supervised single-task learning techniques. Furthermore, the relatedness of tasks may affect the final performance in the multi-task learning framework.
Liu et al. [39] proposed a cross-platform model based on multi-task linear regression. They applied the model to multiple datasets for small interfering RNA (siRNA) efficacy prediction.Given a representation of siRNAs as feature vectors, a linear ridge regression model was applied to predict novel siRNA efficacy from a set of siRNAs with known efficacy. Experiments were run to compare the proposed multi-task learning and traditional single task learning based on root mean squared error. It was shown that in siRNA efficacy prediction, there exists a certain efficacy distribution diversity across the siRNAs bindings onto different mRNAs. Common properties across different siRNAs have influence on potent siRNA design. One way to represent the gene expression data is via a matrix, where gene samples are rows and gene expression conditions are columns.There are two categories of rows: control or case, for any sample.The objective of gene expression classification is to accurately classify new samples based on conditions into case or control. This problem is particularly challenging because the data is very imbalanced: the number of samples are often smaller than the number of features (columns). Furthermore, the data can be very noisy.
Chen et al. [40] proposed a multi-task method, known as support vector sample learning (MTSVSL), and demonstrated that the genes selected by MTSVSL yield superior classification performance when using cancer gene expression data. They started by extracting significant samples that reside on support vectors, and then learned two tasks using multiple back-propagation neural networks simultaneously. They configured their system so that the output of the second task can help refine the original model. One task is aimed at answering “what kind of sample is this,” and the second task is “is this sample a support vector sample?” Their experimental results show that the second task can improve classifier performance by incorporating the generated bias with the original bias obtained from the first task.
In the area of genetic analysis, one of the main issues faced today is GWAS. In this area, Puniyan et al. [41] developed a novel multitask regression based technique to perform joint GWAS mapping on individuals from multiple populations. Initially assuming that there is no population structure, the a lasso function can be formulated as:
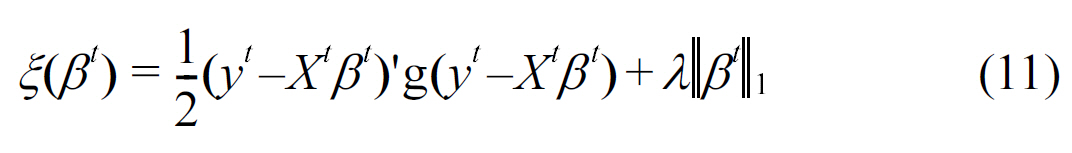
where
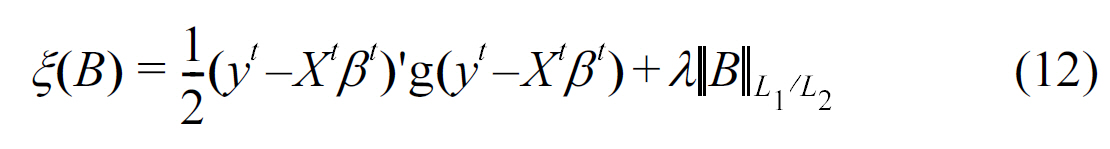
In the above equation,
Both gene expression data analysis and genetic analysis suffer from a lack of sample data due to the high cost of data collection in biology. However, the existence of gene expression data under various platforms or conditions, and genome-wide SNP data of different populations provides us with an opportunity to explore relatedness among multiple datasets to alleviate this data scarcity problem. So far, there have only been a few works in this field [40-42] that use transfer learning methods in machine learning. Future studies are expected to explore how to find the most informative genes or SNPs when classifying different sample conditions by investigating their global characteristics among multiple datasets. This objective can be achieved by regularization-based approaches, learning common feature representation approaches, and distribution matching approaches.
In recent years, the application of transfer learning to systems biology has become increasingly popular. Transfer learning techniques implemented by task regularization, distribution matching, matrix factorization and Bayesian approaches have all been employed in computational systems biology.
Gene interaction network analysis has been very useful for gaining insights into various cellular properties. In 2005,Tamada et al. [43] utilized evolutionary information of two organisms to reconstruct their individual gene networks. Suppose that we have two organisms
As a follow-up, in 2008, Nassar et al. [44] proposed a new score function that captures the evolutionary information shared between
Kato et al. [45] considered multiple assays where learning via the sharing of local knowledge occurs, reflected by learning weights in the formula below:
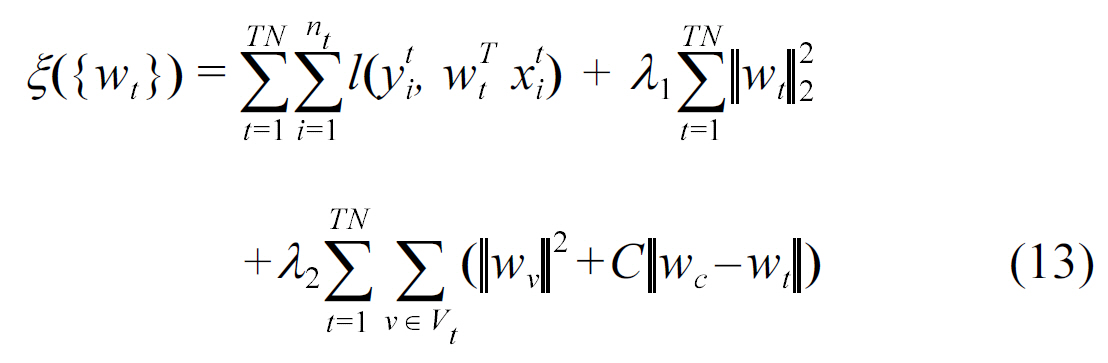
where
Qi et al. [46] proposed a semi-supervised multi-task framework to predict protein-protein interactions from partially labeled reference sets. The basic idea is to perform multitask learning on a supervised classification task and a semi-supervised auxiliary task via a regularization term. This is equivalent to learning two tasks jointly while optimizing the following loss function:
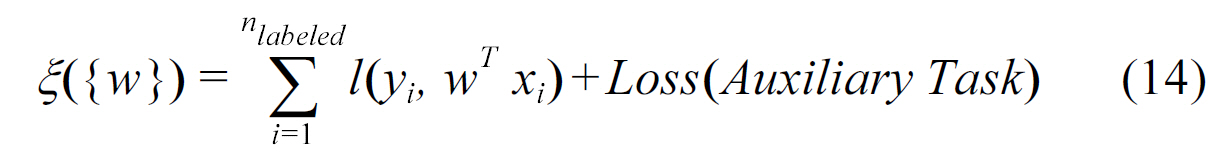
Protein-protein interaction prediction is an important aspect of systems biology. Besides the work of Qi et al. [46], Xu et al.[47] also explored how to use multitask learning in this area via a technique known as Collective Matrix Factorization (CMF)[48]. These methods use similarities of the proteins in two interaction networks as the corresponding shared knowledge. They showed that when the source matrix is sufficiently dense and similar to the target PPI network, transfer learning is effective for predicting protein-protein interactions in a sparse network.Consider a similarity matrix
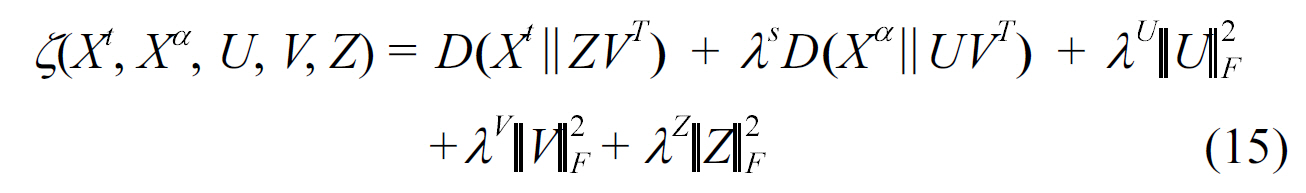
where
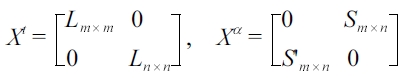
A sparse multitask regression approach was presented in[49], where a co-clustering algorithm is applied to gene expression data with phenotypic signatures. This algorithm can uncover the dependency between genes and phenotypes. A multitask learning framework with
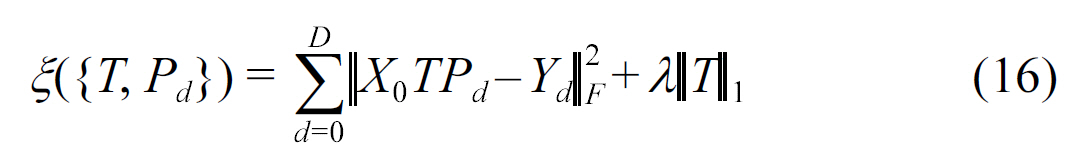
where
Bickel et al. [50] studied the problem of predicting HIV therapy outcomes of different drug combinations based on observed genetic properties of the patients, where each task corresponds to a particular drug combination. They proposed to jointly train models of different drug combinations by pooling data together from all tasks and then use re-sampling weights to adapt the data for each particular task. The goal is to learn a hypothesis
Text mining provides rich ground for transfer learning and multi-task learning applications. In this area, several approaches to general text mining with transfer learning have been surveyed in [3].
In the biomedical domain, one important area is semantic role labeling (SRL) systems, these label the roles of genes, proteins and biological entities discussed in textual form. These texts are often labeled based on manually annotated training instances, which are rare and expensive to prepare. To solve this problem, Dahlmeier and Ng [51] formulated SRL in the biomedical domain as a transfer learning problem, to leverage existing SRL resources from the newswire domain. They employed three domain transfer learning methods: instance weighting, the augment method and instance pruning. Instance weighting [6, 52] is aimed at correcting the probability estimate for the target domain by weighting instances that occur in the auxiliary data sets. Domain adaptation methods [53] map features from the auxiliary and target domains to a common feature space where knowledge transfer is possible.
In addition to biomedical text mining applications of transfer learning, Bi et al. [54] formulated the detection of different types of clinically-related abnormal structures in medical images using multitask learning. Their method captured taskdependence by sharing common feature representations, this was shown to be effective in eliminating irrelevant features and identifying discriminative features. Given
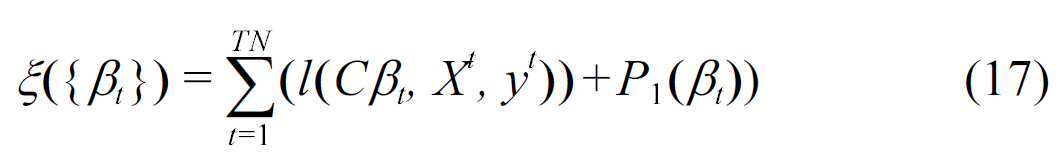
where P1 and P2 are regularization operators, and
In the field of sensor-based ubiquitous healthcare, and particularly in motion-sensor-based activity recognition, collecting user labeled samples needs huge manual efforts and may involve privacy issues. Therefore, transfer learning becomes an attractive approach to solving the data sparsity problem. Here we give one example of such a work, which transferred the activity models for a new user by transfer learning [55]. In earlier work, transfer learning was employed to transfer the activity models learned for a user to another user. Van Kasteren et al.[56] described a simple method for transferring the transitional probabilities of two Markov models for two different spaces. In this work, the aim is to recognize activities in a new space without the expensive data annotation and learning processes. Kasteren et al. [56] first mapped data between two sensor networks via a mapping function. Then their algorithm learned parameters using the labeled data in the source space and unlabeled data in the target space. In this work, the mapping is generated manually and the structure of the HMMs is pre-defined. However,in practice, the mapping and model structure should be learned.
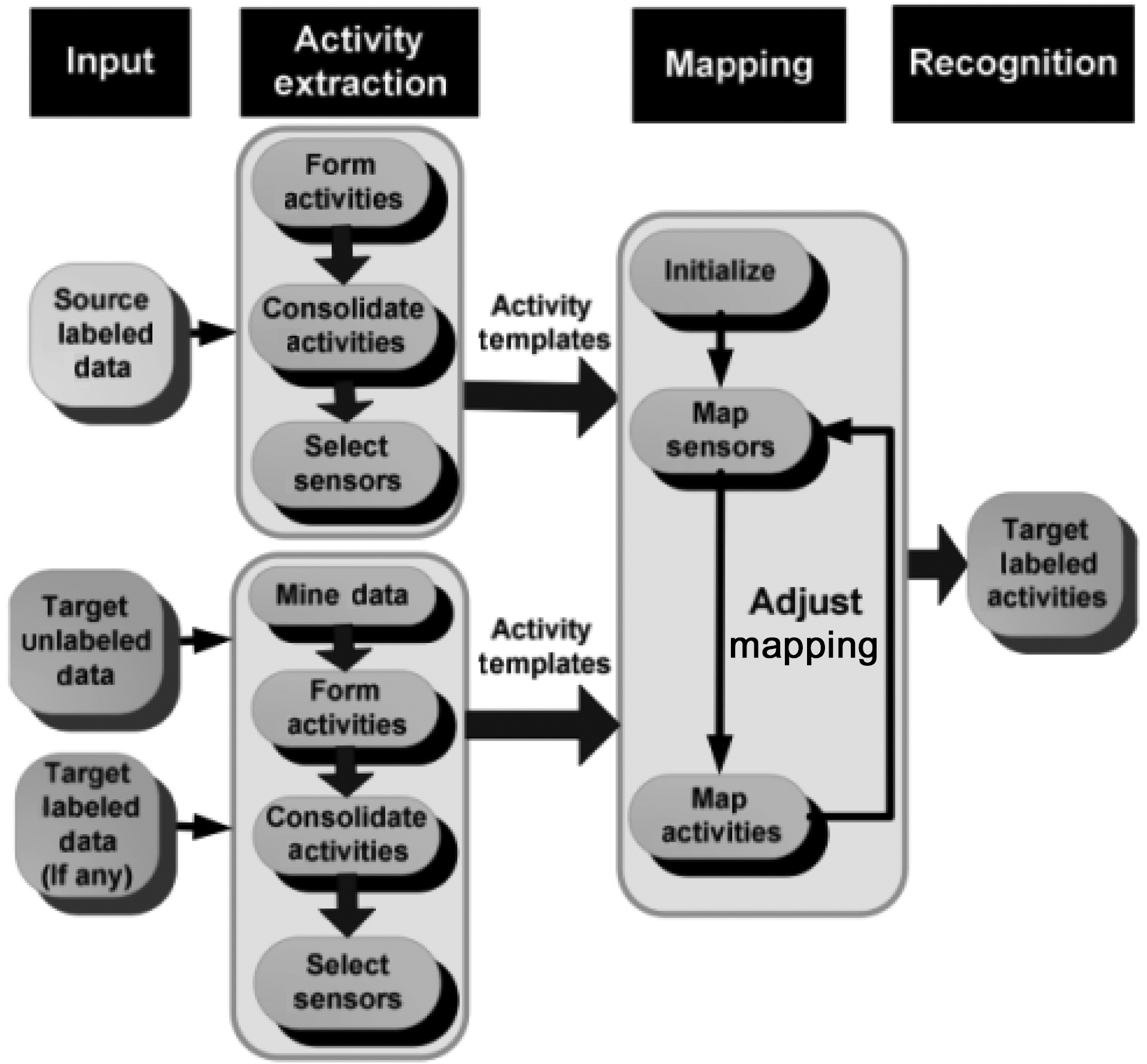
To address this problem, Rashidi and Cook [57] proposed an advanced transfer learning approach to transfer activity knowledge learned in a home to another home, in what they call“Home to Home Transfer Learning” (HHTL). The main components of HHTL are illustrated in the Fig. 4. As the figure shows,HHTL extracts and compresses activity models from the sensor
data based on label information in a source domain where data is assumed to have been collected and labeled. This data can then be used to build activity models that transfer to the target domain. After extracting activities and their corresponding sensors,source activity models are mapped to target activity models using a semi-EM framework in an iterative manner. Initially,sensor mapping probabilities are adjusted based on activity mapping probabilities. Next, the activity mapping probabilities are adjusted based on the updated sensor mapping probabilities.A target activity’s label is determined to be the same as the source activity’s label when the mapping probability is maximized.
Subsequently, Rashidi and Cook [58] modified the HHTL algorithm to transfer the knowledge of learned activities from multiple source physical spaces. E.g., knowledge transfer is done from two homes A and B, to a target physical space, e.g.home C. The modified learning model (MHTL) has the same first three steps illustrated in Fig. 4, but in the final step, MHTL adopts a weighted majority voting scheme to assign the final labels to activities in the target domain.
VI. DATA SETS FOR TRANSFER LEARNING
Several datasets have been released for further research on transfer learning in biological domains. We list several datasets available here.
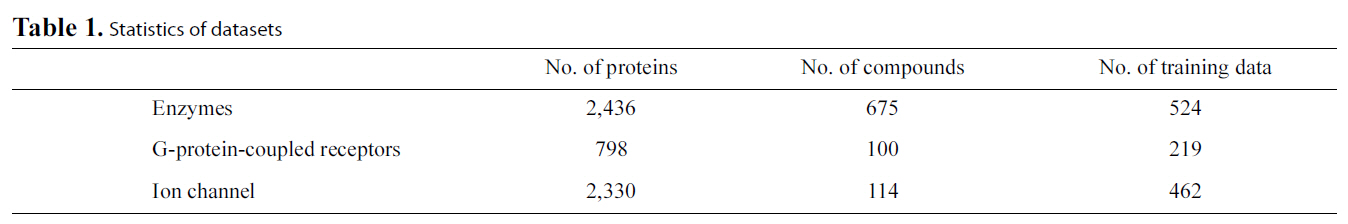
[Table 1.] Statistics of datasets
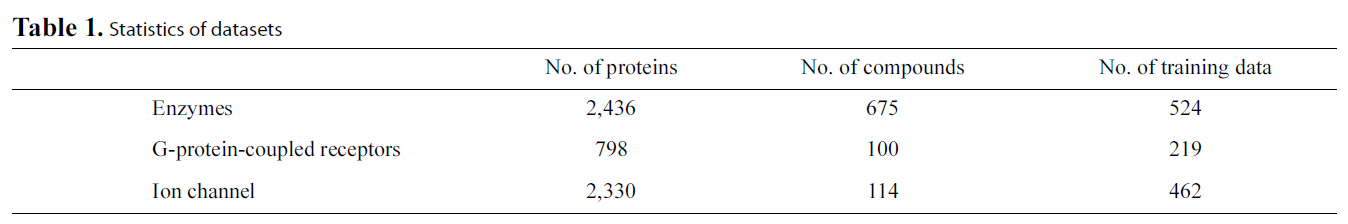
Statistics of datasets
Gene Expression Data: Yeast gene expression data and human gene expression data are available from ([59], http://genome-www.stanford.edu/cellcycle/) and ([60], http://www.stanfore.edu/Human-CellCycle/Hela/), respectively. These two sets of gene-expression data can be used to reconstruct gene networks across different organisms.
Protein-ligand Binding Data: One group of available data is composed of enzyme-ligand interaction data, G-protein-coupled receptors (GPCR)-ligand interaction data and ion channelligand interaction data (http://bioinformatics.oxfordjournals.org/content/24/19/2149/suppl/DC1). Table 1 lists the statistics of the data. Another group of released ligand interaction data contains four subsets for enzyme, ion channel, GPCR and nuclear receptors [61] (http://web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/),respectively. The ligand structure similarity matrices and protein sequence similarity matrices are also included in these datasets. Interested readers can build models to predict the protein-ligand binding via a multi-task framework using these datasets or predict the protein-ligand binding via transferring knowledge from auxiliary data such as the ligand structure similarity matrices and protein sequence similarity matrices.
HIV-1 and Human Interaction Data: Qi et al. [46] preprocessed a HIV-1 and protein interaction dataset (http://www.cs.cmu.edu/qyj/HIVsemi/), and exploited a semi-supervised multitask method to predict HIV-1 and protein interactions.

![Illustrating source and target domains in biology (adopted from invited talk by Gunnar Ratsch Invited Talk at NIPS Transfer LearningWorkshop December 2009 Whistler B.C. (Schweikert et al. [31]).](http://oak.go.kr/repository/journal/10861/E1EIKI_2011_v5n3_257_f001.jpg)
![Four domain adaptation models (adopted from invited talk byGunnar Ratsch Invited Talk at NIPS Transfer Learning Workshop December2009 Whistler B.C. (Schweikert et al. [31]).](http://oak.go.kr/repository/journal/10861/E1EIKI_2011_v5n3_257_f002.jpg)
![Main components of Home to Home Transfer Learning (HHTL)for transferring activities from a source space to a target space (Rashidiand Cook [57]).](http://oak.go.kr/repository/journal/10861/E1EIKI_2011_v5n3_257_f004.jpg)