Recently, natural products derived from marine resources have attracted great attention of researchers in drug discovery. Marine brown algae (division Phaeophyta)produce a wide variety of secondary metabolites,such as terpenoids, oxylipins, phlorotannins, volatile hydrocarbons, and steroids. Several of these compounds have been reported to exhibit a wide spectrum of biological activities such as cytotoxic, antimicrobial, and antioxidant activities as well as inhibitory activity against various enzymes (Chang et al. 2008).
In the course of the phytochemical study of the marine seaweeds from Jeju Island, we have now investigated the constituents of
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a JEOL JNM-ECP 400 NMR spectrometer (JEOL, Tokyo, Japan), using dimethylsulfoxide (DMSO)-
The marine brown seaweed,
The dried
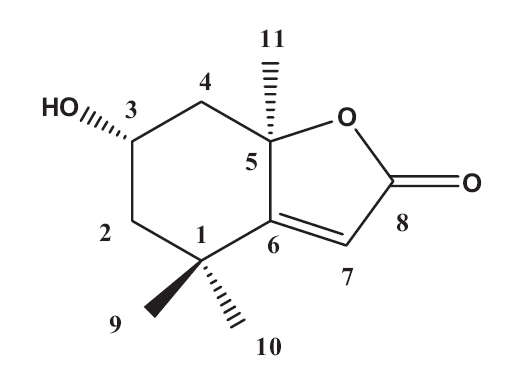
Loliolide: white powder. EIMS:
>
DPPH radical scavenging assay
DPPH radical scavenging activity of the isolated compound was determined according to a slightly modified method of Blois (1958). DPPH solution was prepared at the concentration of 4 × 10-4 M in MeOH. The different concentrations of the test samples (100 μL) and 100 μL of freshly prepared DPPH solution were thoroughly mixed. The reaction mixture was incubated at room temperature for 30 min. Thereafter, the absorbance was measured at 517 nm by enzyme-linked immunosorbent assay (ELISA) reader. The scavenging activity was calculated according the following equation:
DPPH scavenging activity (%) = [1 ? (A sample / A control)] × 100
>
Hydrogen peroxide scavenging activity
Hydrogen peroxide scavenging activity was determined according to the method of Muller (1985). A hundred microliter of 0.1 M phosphate buffer (pH 5.0) and the sample solution were mixed in a 96 microwell plate. A 20 μL of hydrogen peroxide was added to the mixture, and then incubated at 37°C for 5 min. After the incubation, 30 μL of 1.25 mM ABTS and 30 μL of peroxidase (1 unit mL-1) were added to the mixture. The reaction mixture was incubated at 37°C for 10 min. The absorbance was measured at 405 nm by ELISA reader.
Monkey kidney fibroblast cell line (Vero cell) was purchased from Korean Cell Line Bank (KCLB), maintained at 37°C in an incubator with humidified atmosphere of 5% CO2. Cells were cultured at a concentration of 1 × 105 cells mL-1 in Dulbecco’s modified Eagle’s medium containing 10% heat-inactivated fetal calf serum, streptomycin (100 μg mL-1), and penicillin (100 unit mL-1) for further experiments.
>
Determination of intracellular ROS by DCFH-DA
For the detection of intracellular ROS, the DCFH-DA method as previously described by Rosenkranz et al. (1992) was used. Vero cells were seeded in 96-well plates at a concentration of 1 × 105 cells mL-1. After 16 h, the cells were treated with various concentrations of the test samples, and incubated at 37°C under a humidified atmosphere. After 30 min, 10 μL of 1 mM H2O2 was added, and then cells were incubated for an additional 30 min at 37°C. Finally, 2′,7′-dichloro-dihydrofluorescein diacetate (DCFH-DA; 5 μg mL-1) was introduced to the cells, and 2′,7′-dichlorodihydrofluorescein fluorescence was detected at an excitation wavelength of 485 nm and an emission wavelength of 535 nm, using a Perkin-Elmer LS-5B spectroflurometer (Waltham, MA, USA). The percentage of intracellular ROS scavenging activity was calculated according to the following equation:
Intracellular ROS scavenging activity (%) = [1 ? (C1 / C0)] × 100
Where C1 is the fluorescence intensity of cells treated with H2O2 and the test samples, and C0 is the fluorescence intensity of cells treated with H2O2 and distilled water instead of the test samples.
>
Assessment of cell viability
Cell viability was estimated via a MTT assay, which is a test of metabolic competence predicated upon the assessment of mitochondrial performance. It is a colorimetric assay, which is dependent on the conversion of yellow tetrazolium bromide to its purple formazan derivative by mitochondrial succinate dehydrogenase in viable cells (Mosman 1983). The cells were seeded in 96-well plate at a concentration of 1 × 105 cells mL-1 (180 mL). After 16 h incubation at 37°C under a humidified atmosphere, the cells were treated with various concentrations of the compound (10 μL), and further incubated for 30 min. Then, 10 μL of H2O2 (1 mM) was added to the cell culture medium, and incubated for 24 h at 37°C. The kr50 μL of MTT stock solution (2 mg mL-1) was then applied to the wells, to a total reaction volume of 250 mL. After 4 h of incubation, the plates were centrifuged for 5 min at 800 ×g, and the supernatants were aspirated. The formazan crystals in each well were dissolved in 150 mL of DMSO, and the absorbance was measured via ELISA at a wavelength of 540 nm. Relative cell viability was evaluated in accordance with the quantity of MTT converted to the insoluble formazan salt. The optical density of the formazan generated in the control cells was considered to represent 100% viability. The data are expressed as mean percentages of the viable cells versus the respective control.
>
Nuclear staining with Hoechst 33342
The nuclear morphology of the cells was evaluated using the cell-permeable DNA dye, Hoechst 33342. Cell with homogeneously stained nuclei were considered viable, whereas the presence of chromatin condensation and / or fragmentation was indicative of apoptosis (Gschwind and Huber 1995, Lizard et al. 1995). The cells were placed in 24-well plates at a concentration of 1 × 105 cells mL-1 (950 μL). Sixteen h after plating, the cells were treated with various concentration of the compounds (50 μL), and further incubated for 1 h prior to expose to H2O2 (1 mM). After 24 h, 3 μL of Hoechst 33342 (stock 10 mg mL-1), a DNA-specific fluorescent dye, were added to each well, followed by 10 min of incubation at 37°C. The stained cells were then observed under a fluorescence microscope equipped with a CoolSNAP-Pro color digital camera (Maryland, USA), in order to examine the degree of nuclear condensation.
Cell cycle analysis was conducted to determine the proportion of apoptotic sub-G1 hypodiploid cells (Nicoletti et al. 1991). The Monkey kidney fibroblast cells (Vero cell) were plated in 6-well plates at a concentration of 4.0 × 105 cells mL-1 and incubated for 24 h at 37°C under a humidified atmosphere. The cells were then treated with different concentration of loliolide. After incubation for 1 h, 150 μL of 1 mM H2O2 was added, and the cells were incubated for an additional 6 h at 37°C. Finally the cells were harvested and fixed in 1 mL of 70% ethanol for 30 min at 4°C. The cells were washed twice with phosphate buffered saline (PBS) and incubated in darkness in 500 μL of PBS containing 50 μg PI and 50 μg RNase A for 30 min at 37°C. Flow cytometric analysis was conducted
with a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). The effect on the cell cycle was determined by changes in the percentage of cell distribution at each cell cycle phase, and assessed by histograms generated by the Quest and Mod-Fit computer programs (Wang et al. 1999).
>
Isolation and structure determination of loliolide from Sargassum ringoldianum subsp. coreanum
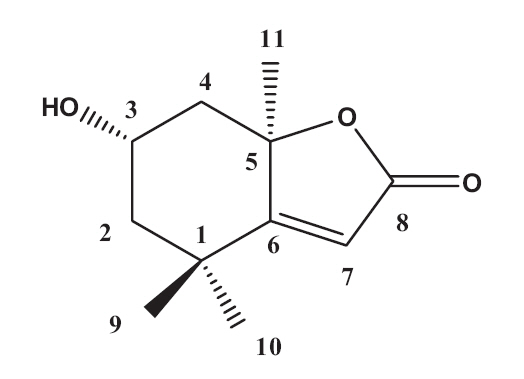
The EtOAc fraction was purified by silica gel and ODS column chromatography (Fig.1 ). A monoterpenoid compound, loliolide, was successively isolated using HPLC from the sub-fraction. The chemical structure of the compound was elucidated on the basis of spectral analysis of MS and NMR (1H and 13C) data, and also comparing with the previous published data (Zhang et al. 2010). The compound, loliolide, was obtained from this species for the first time. The structure was shown in Fig. 2.
>
Antioxidant activities of loliolide
To evaluate antioxidant activities of loliolide isolated from
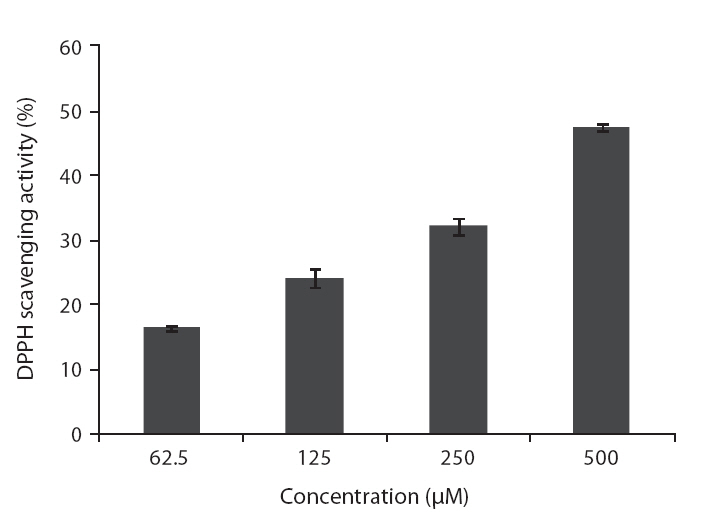
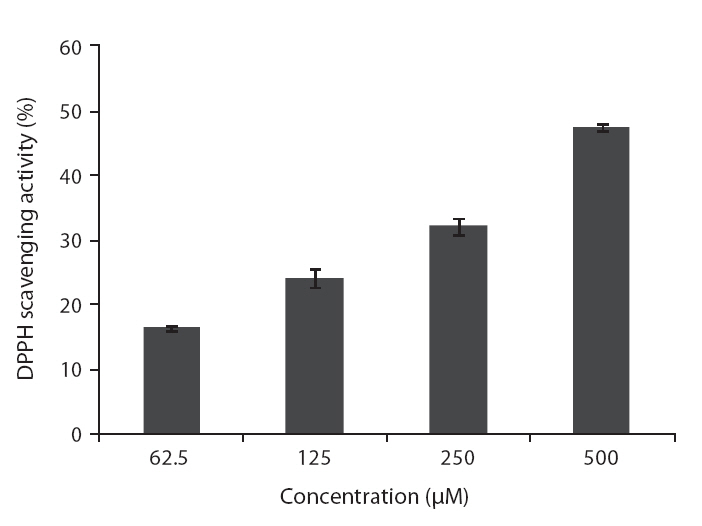
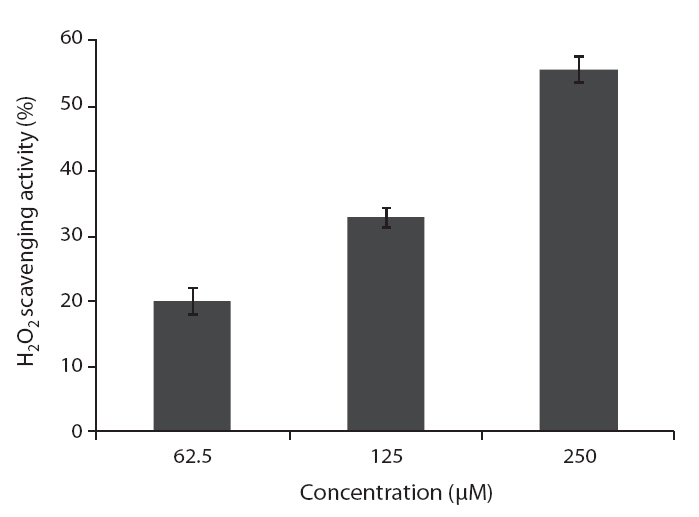
DPPH is a free-radical generating compound and has been widely used to evaluate the free-radical scavenging ability of various samples. It is a stable free radical and accepts an electron or hydrogen radical to become a stable diamagnetic molecule (Li et al. 2009). Hydrogen peroxide is a product derived from normal metabolism, but is not an inherently reactive non-radical compound. However, hydrogen peroxide can be converted into highly reactive and deleterious products such as oxygen, and hydroxyl radical (Heo et al. 2005
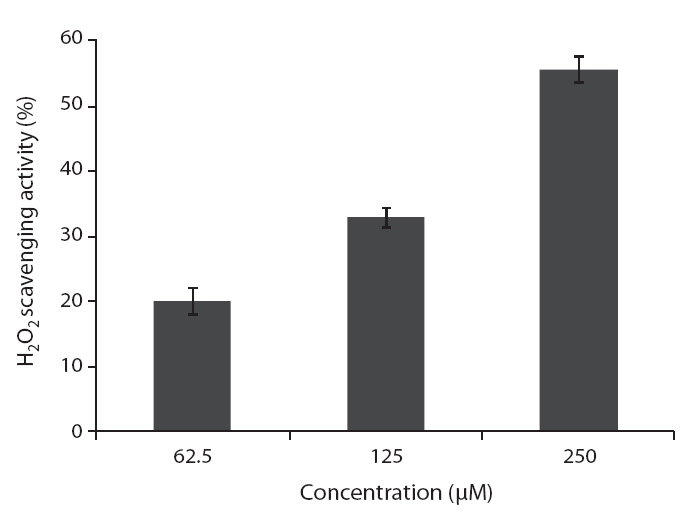
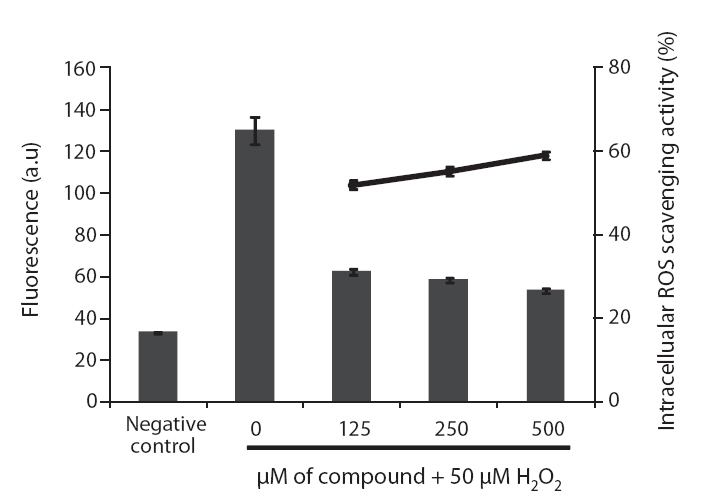
ROS scavenging activities of loliolide was 52.0, 55.3 and 59.1% at concentration of 125, 250, and 500 μM, respectively (Fig.5 ).
>
Determination of cell protective effects of loliolide
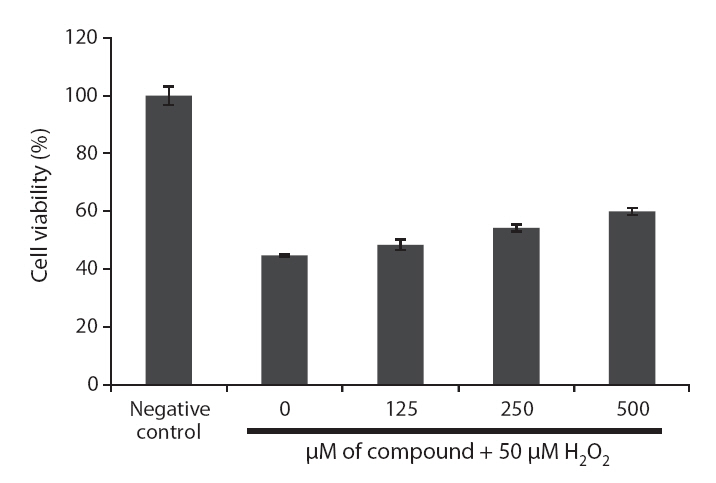
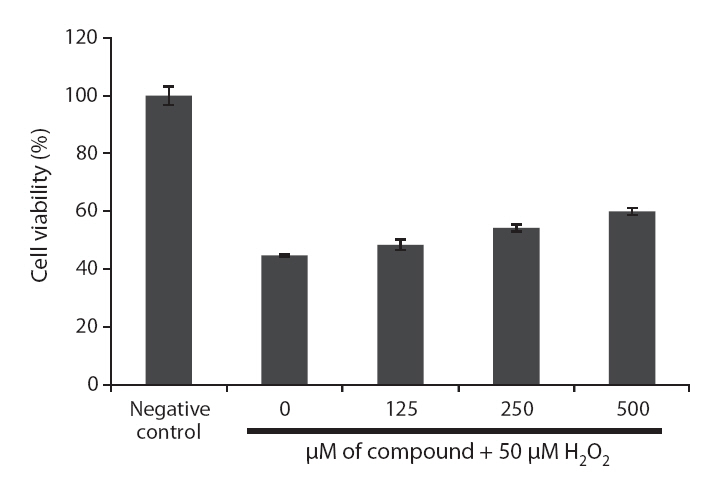
Because loliolide showed DPPH free radical, H2O2 and intracellular ROS scavenging activity, the cell protective effects of loliolide on H2O2-induced cell damage were also determined via different assays, including MTT, nuclear staining and cell cycle analysis. The protective effect of loliolide on cell viability against H2O2-induced Vero cell damage was assessed using MTT assay. As shown in Fig. 6,cell viability in non-treated cell was assigned as 100%, whereas in the H2O2-treated cells, the cell viability was reduced to 44.8%. However, loliolide could prevent the H2O2-induced cell damage. The cell viability was increased to 48.4, 54.3 and 60.0%, as increasing the concentrations from 125 to 500 μM.
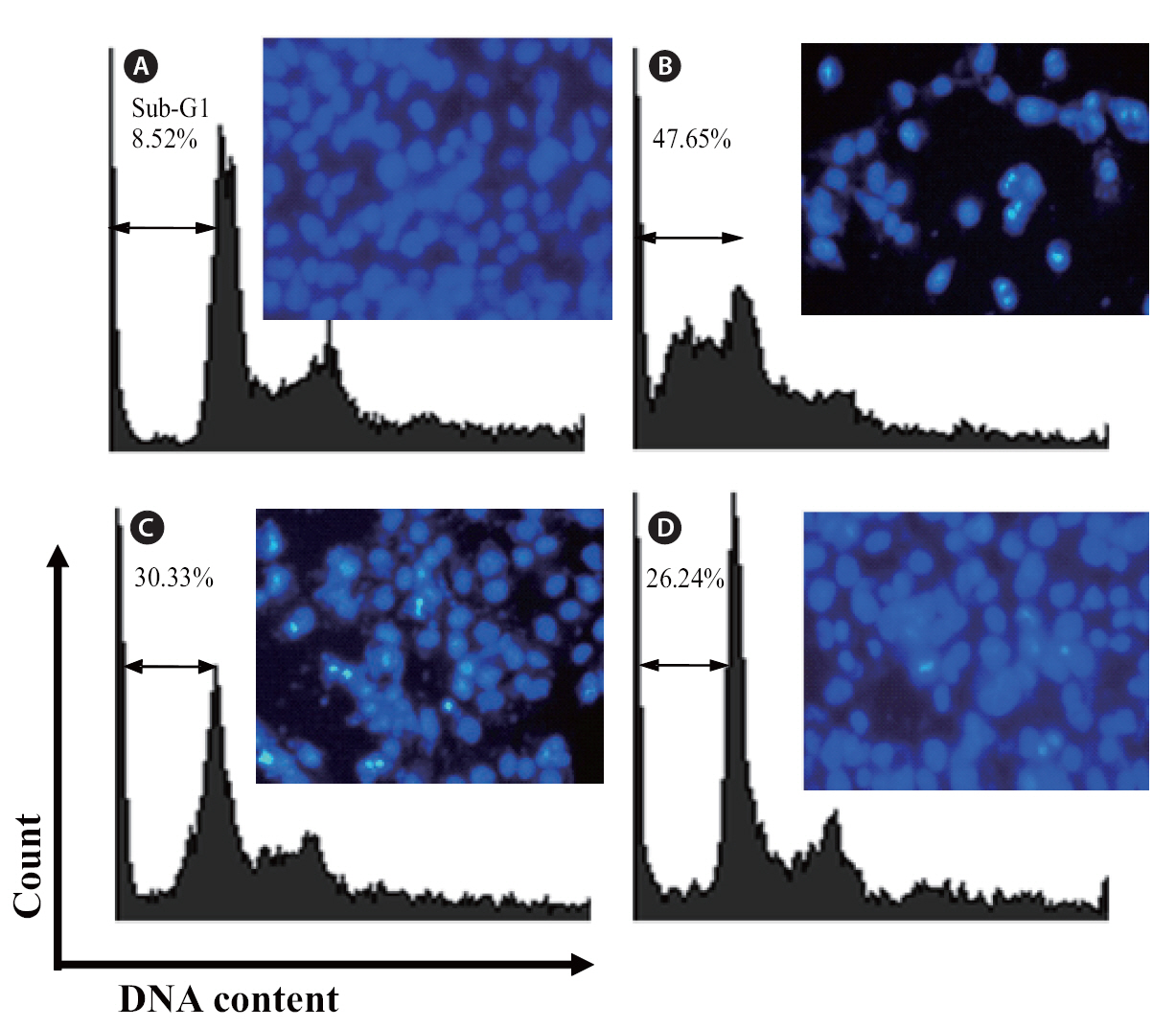
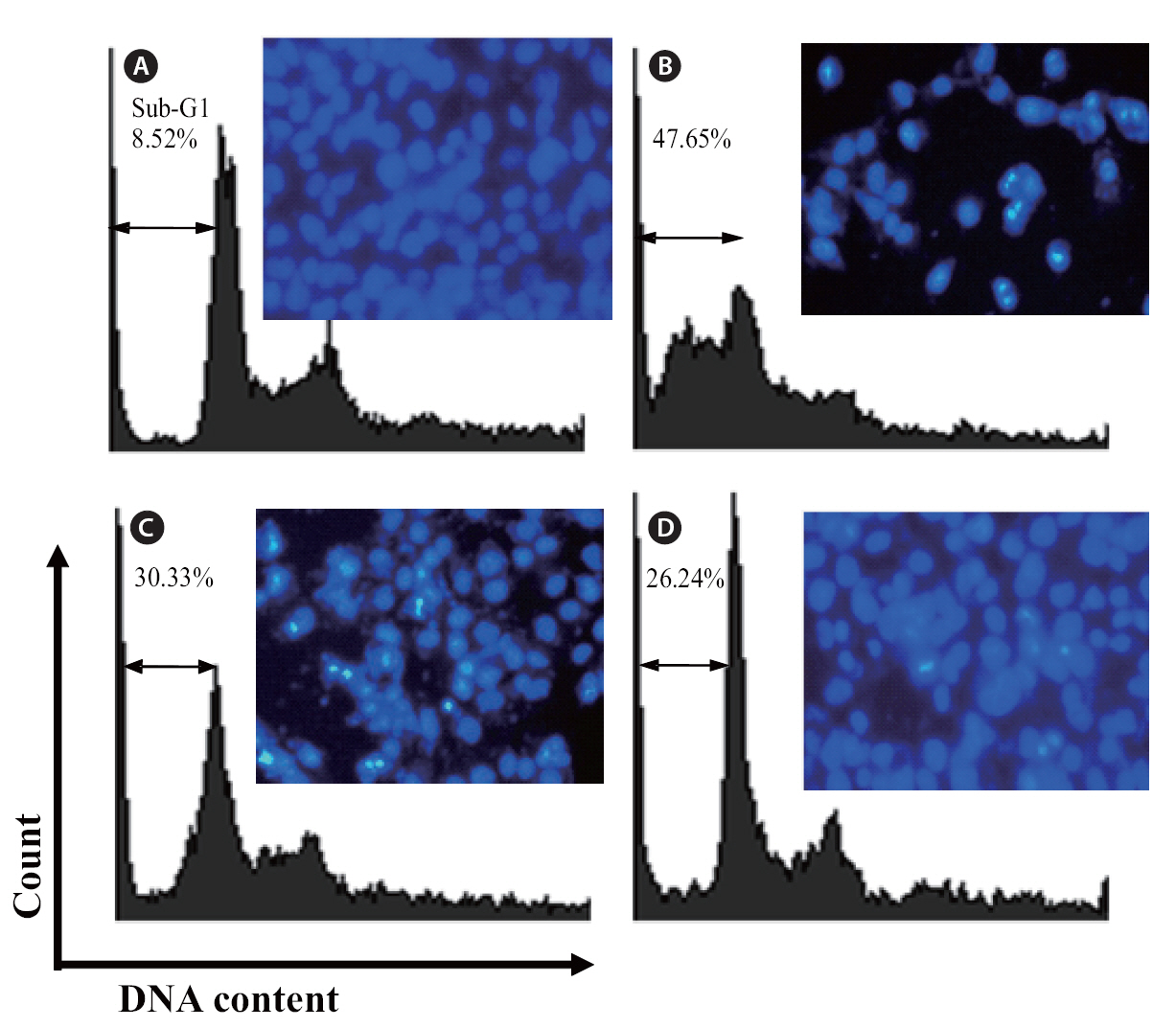
The protective effects of loliolide on H2O2-induced apoptosis were investigated via nuclear staining with Hoechst 33342. Hoechst 33342 is used for specifically staining the nuclei of living or fixed cells and has been used extensively for the detection of nuclear shrinkage, including chromatin condensation, nuclear fragmentation, and the appearance of apoptotic bodies, all of which are indicative of apoptosis. As shown in Fig. 7,uninjured nuclei were clearly observed in the non-treated group (Fig.7 A). When the cell was treated with H2O2, cell apoptosis which was expressed as a significant level of nuclear fragmentation was appeared (Fig.7 B). However, the cells co-incubated with different concentration of loliolide and H2O2 could reduce the apoptosis of cells (Fig.7 C & D). Particularly, at the concentration of 500 μM, a significant reduction of fragmentation was observed. This result suggested that loliolide show an ability to protect cells against H2O2-induced nuclear fragmentation.
Finally, the protective effects of loliolide were also investigated via flow cytometry. Compared with non-treated cells (Fig 7A), the sub-G1 DNA content was increased to 47.5% when the cells were treated with H2O2 (Fig. 7B). This result revealed that addition of H2O2 could induce the apoptosis. Whereas, the cells pretreated with 250 and 500 μM of loliolide, the sub-G1 contents were significantly reduced to 30.33 and 26.24%, respectively.
Recently, marine brown and red algae have been well-
known as an important source of bioactive components. Many reports showed that these components exhibited various bioactivities. Marine brown alga,
ROS including superoxide anion (?O2-), hydroxyl radical (?OH), peroxyl (ROO?), alkoxyl (RO?), nitric oxide and hydrogen peroxide (H2O2) is thought to be one of the causes of chronic diseases such as heart disease, inflammation, aging, and cancer. ROS can be produced by endogenous sources such as normal aerobic respiration, simulated polymer phonuclear leukocytes and macrophages, peroxisomes, and exogenous sources such as smoking, ionizing radiation, certain pollutants, organic solvents, pesticides, and a high polyunsaturated fatty acid diet. Among the ROS, H2O2 is one of the most crucial as it can generate other species of ROS, such as hypochlorous acid (HOCl), via the enzymatic (myeloperoxidase) oxidation of chloride ions. HOCl can also result in the production of highly reactive singlet oxygen or even hydroxyl peroxycarbonate. Finally, these ROS could react with bimolecular substance
According to our knowledge, there is no literature regarding to the antioxidant activity of loliolide. This is first report focusing on the antioxidant properties of loliolide isolated from
As loliolide showed antioxidant effects against DPPH, H2O2 radicals and intercellular ROS, the cell protective effects of loliolide against the cellular damage induced by H2O2 were also evaluated. The cell protective of loliolide against H2O2-induced cell death or damage can be observed by enhance the cell viability. Furthermore, the morphology changes regarding to the cell protective effect was also investigated via nuclear staining with Hoechst 33342. Hoechst 33342 is used for specifically staining DNA, and has been used extensively for the detection of nuclear shrinkage, including chromatin condensation, nuclear fragmentation, and the appearance of apoptotic bodies, all of which are indicative of apoptosis. High ROS level in the cell could lead to apoptosis and necrosis, both of which processes have been implicated in cancer, aging, heart diseases and neurodegenerative disorders. In this study, the obvious apoptosis can be observed when the cell was challenged with H2O2. H2O2 are able to induce damage to various biological macromolecules, including DNA, RNA, proteins and lipids. Thus it is very important to study the substances that scavenge H2O2. Our results showed that the treatment of different concentration of loliolide was found to significantly reduce the nuclear fragmentation in the H2O2-treated cells. Finally, the cell protective effects of loliolide associated with influence of cell cycle arrest in G1 phase were also studied. Treatment with different concentration of loliolide in H2O2-treated Vero cells could reduce the sub-G1 DNA contents.
In conclusion, a monoterpenoid, loliolide isolated from