이 연구는 봉약침의 봉독이 NF-κB 활성억제와 안드로겐 수용체 조절 단백질 및세포자멸사 조절 단백질의 발현을 통하여 세포자멸사를 유도하고, 전립선 암세포를 이식한쥐에서의 세포자멸사 유도 효과를 확인함으로써, 봉약침의 봉독이 생체 내에서도 세포자멸사를 유도하여 전립선암에 효과를 나타냄을 확인하고자 하였다.
세포자멸사의 관찰에는 DAPI, TUNEL staining assay를 시행하였으며, 세포자멸사 조절 단백질의 변동 관찰에는 western blot analysis를 시행하였고, 세포자멸사와 연관된NF-κB의 활성 변화를 관찰하기 위해 EMSA를 시행하였다.
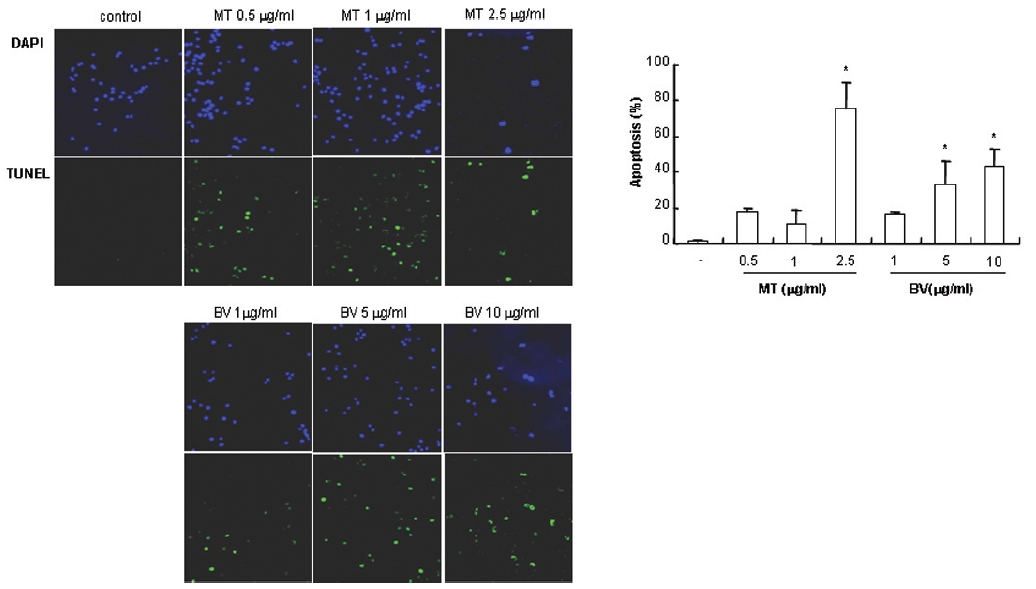
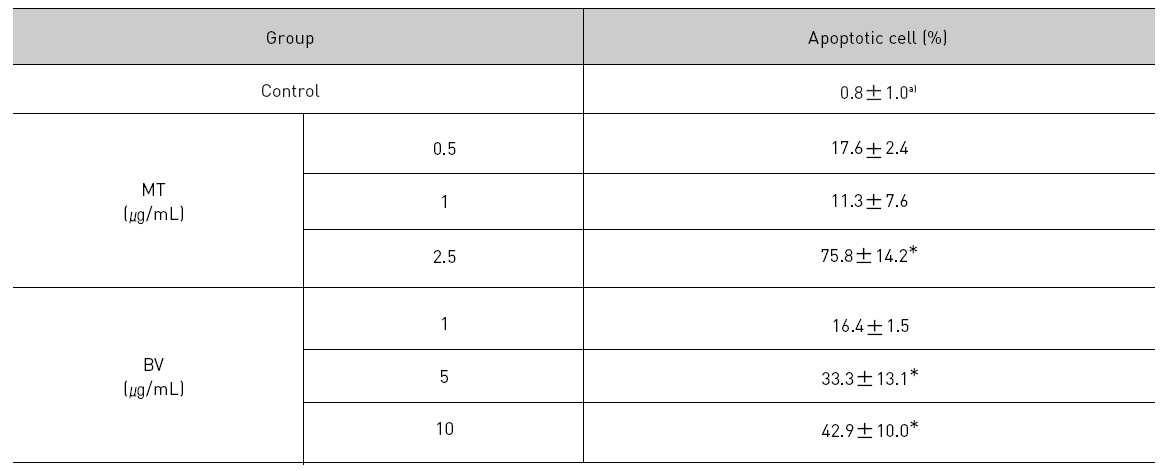
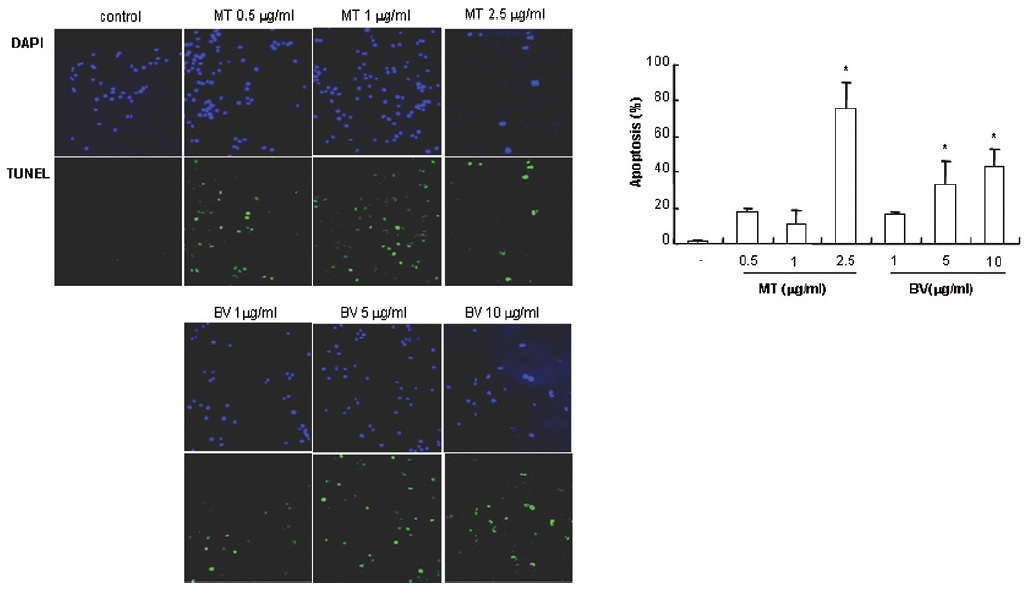
1. DAPI, TUNEL staining assay 결과 봉독 및 melittin을 처리한 LNCaP 세포 모두에서 세포자멸사 유도율이 유의한 증가를 나타내었다.
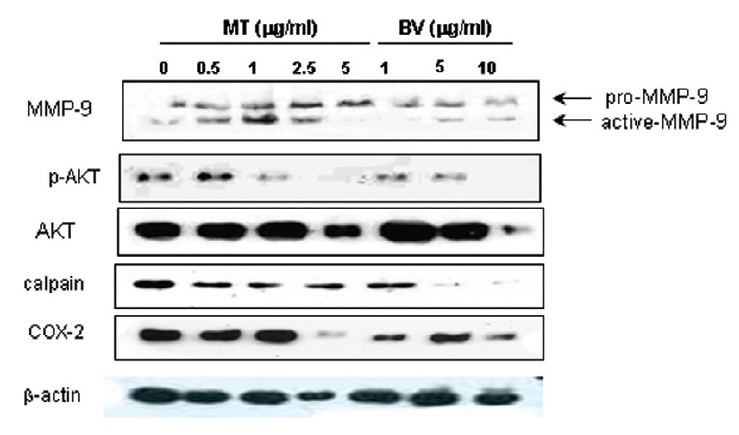
2. LNCaP 세포에 봉독이나 melittin을 처리한 결과, 안드로겐 수용체 조절 단백질 중 p-Akt, COX-2, calpain은 봉독과 melittin 모두에서 유의한 감소를 나타내었고, Akt는 melittin에서유의한 감소를 나타냈으며, 봉독에서 증가하는 경향을 보였고, MMP-9은 증가하였다.
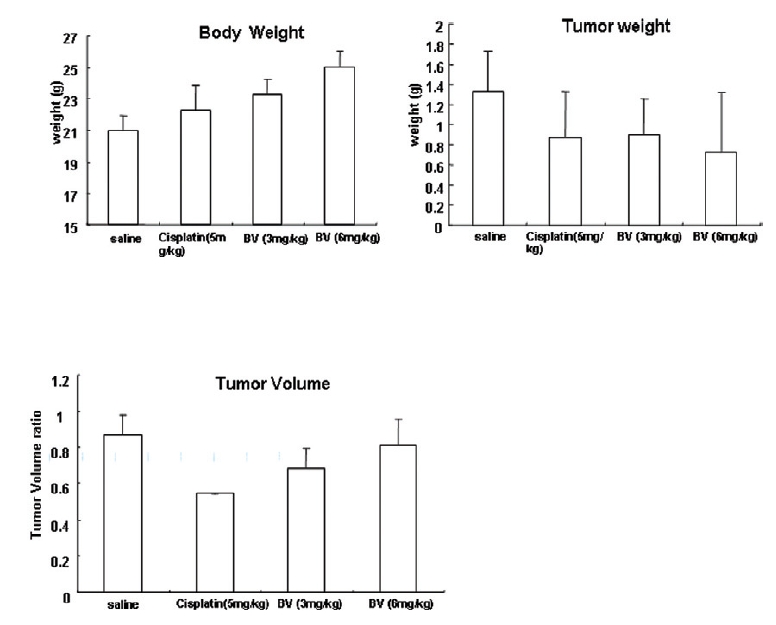
3. 생체 내에서의 봉독의 항암효과를 확인하기 위해 전립선암세포가 이식된 쥐에 봉독을처리한 후 암세포의 부피와 무게, 쥐의 체중을 측정한 결과, 봉독을 처리한 군에서 암세포 부피비율 및 무게는 감소하였고, 쥐의 체중은 증가하였다.
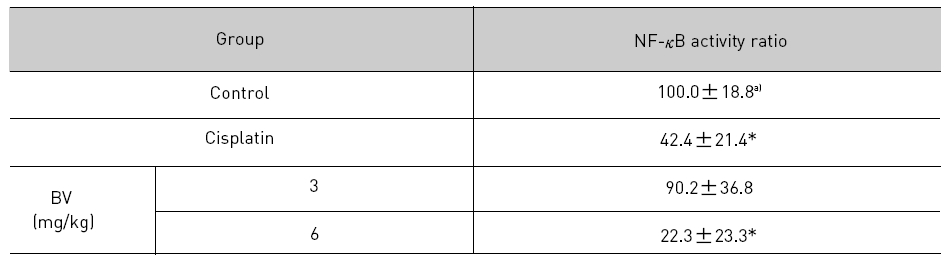
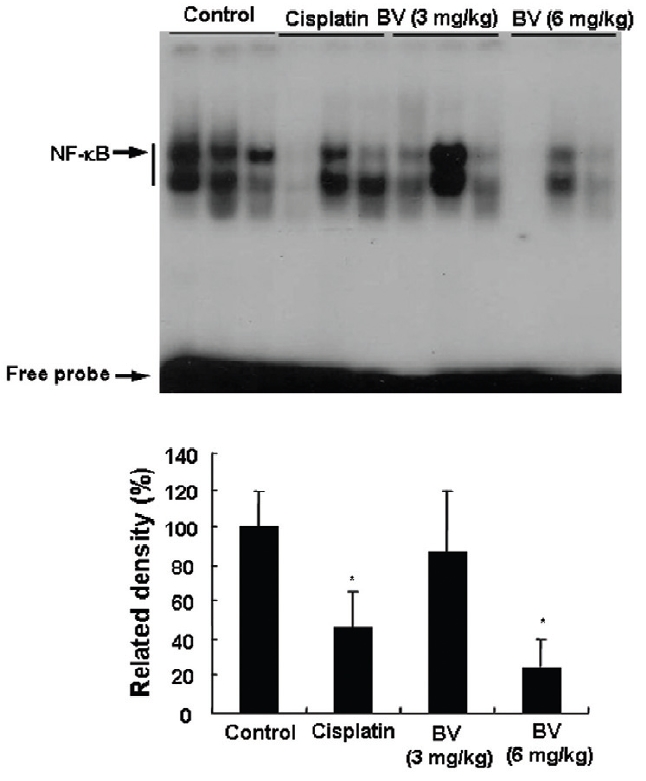
4. 전립선암세포가 이식된 쥐에 봉독을 처리한 결과, NF-κB 활성에서 유의한 감소를 나타내었다.
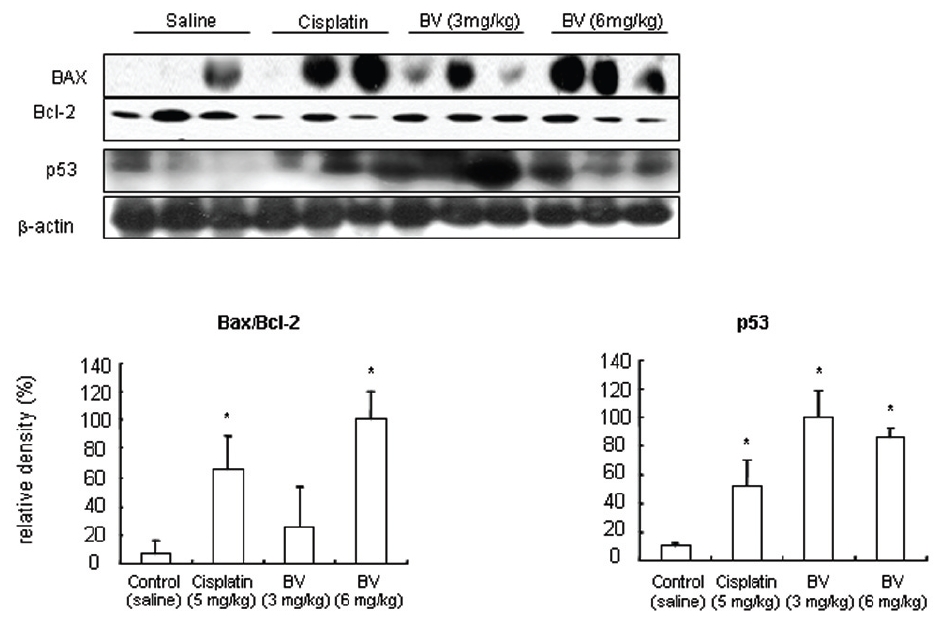
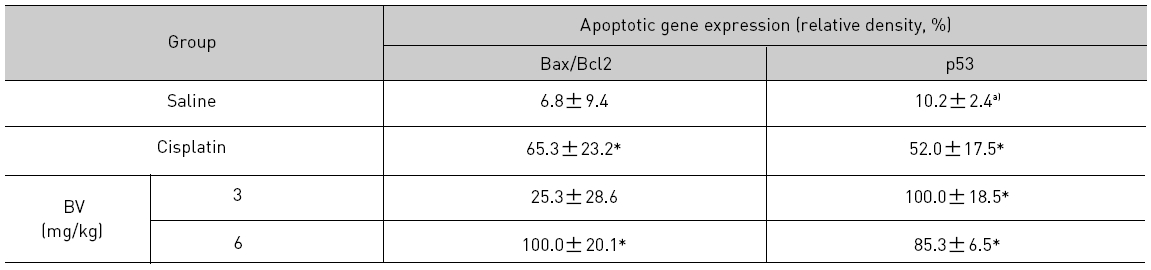
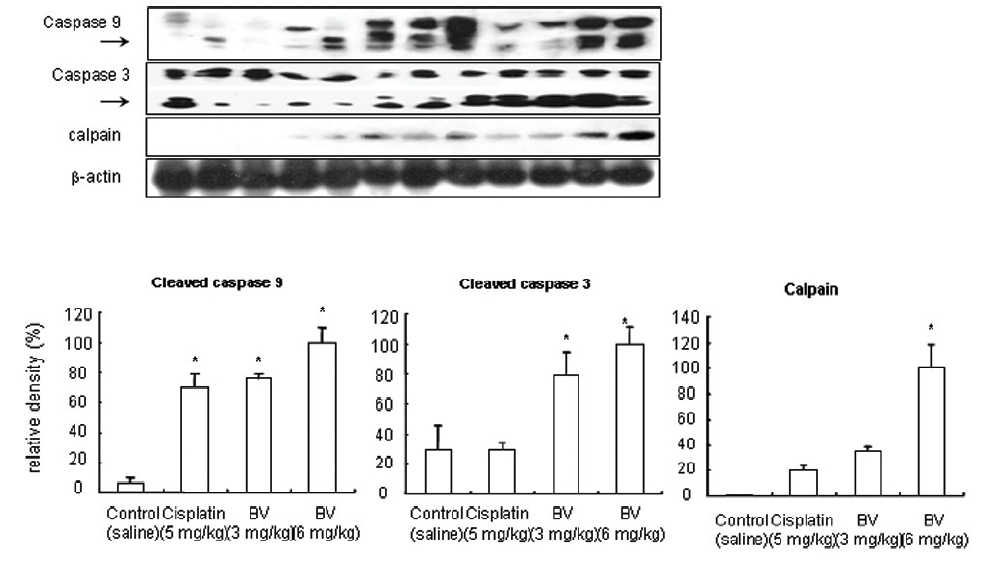
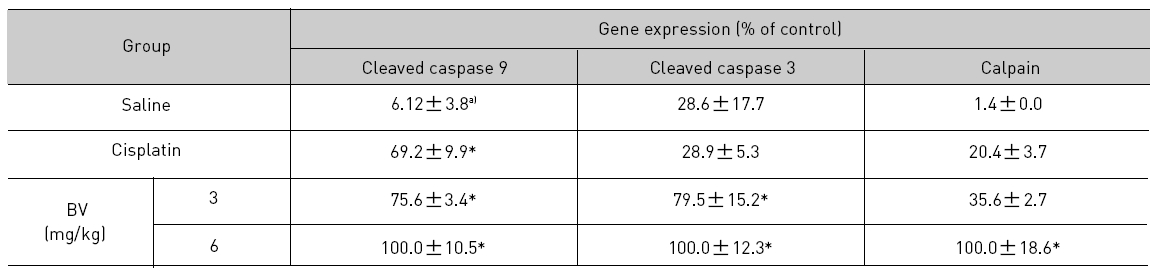
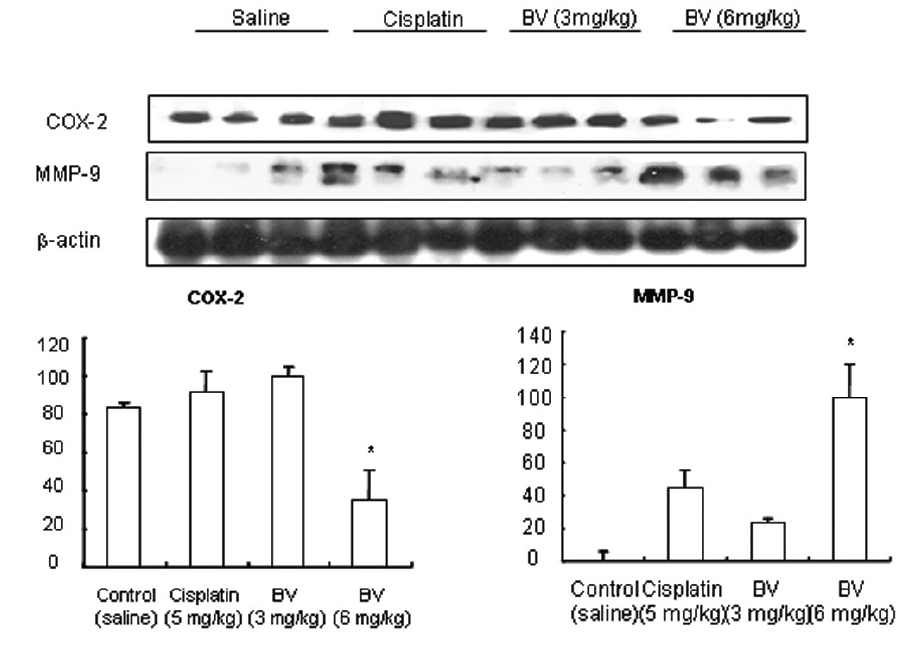
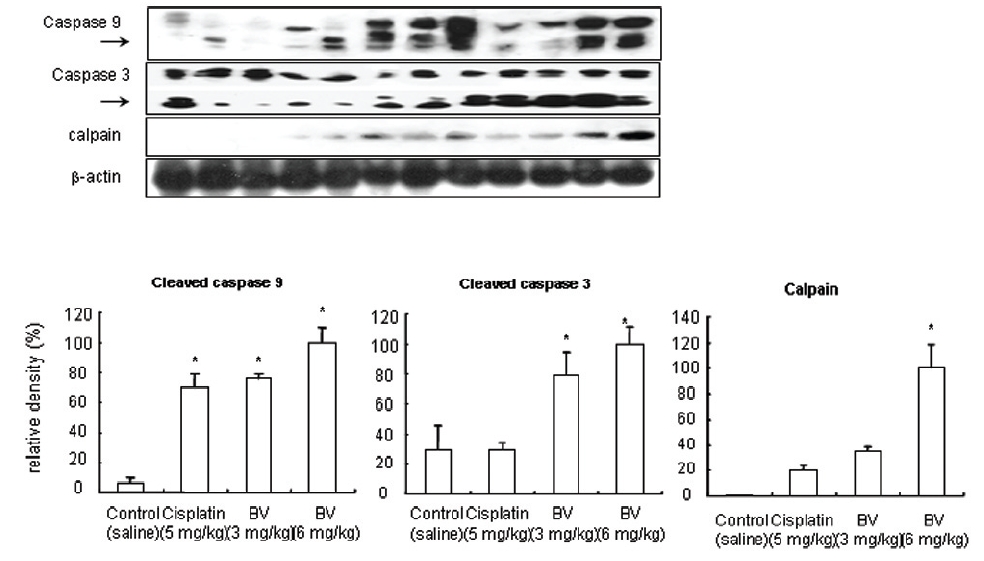
5. 전립선암세포가 이식된 쥐에 봉독을 처리한 결과, 세포자멸사 조절 단백질 중 Bax/Bcl-2, p53, caspase-3, caspase-9, calpain은 유의한 증가를, COX-2는 유의한 감소를 나타냈으며, MMP-9는 증가를 나타내었다.
이상의 결과는 봉독이 시험관 내에서 뿐만 아니라 생체 내에서도 NF-κB의 활성을억제하고 안드로겐 수용체 조절 단백질 및 세포자멸사 조절 단백질의 조절을 통하여 인간전립선암 세포주인 LNCaP의 세포자멸사를 유도함으로써 전립선암 세포 증식억제 효과 및호르몬 비의존적인 전립선암으로의 전이를 지연시키는 경향이 있을 것으로 사료되고, 봉독이 전립선암의 예방과 치료에 효과적으로 활용될 수 있을 것으로 기대된다.
Adenocarcinoma of the prostate (CaP) is the most frequently diagnosed solid tumor in American men and the second leading cause of cancer-related death among men in the United States1). The Well managing prostate cancer remains a complex and intractable problem. Despite of the current existing treatment options available including androgen ablation with luteinizing hormone-releasing hormone agonists, nonsteroidal androgen receptor antagonists, androgen withdrawal, corticosteroids, chemotherapy, radiation therapy, radiopharmaceuticals, immunologic approaches, gene therapy, and novel molecular targets2), metastatic hormone refractory prostate cancer is yet threatening lives as a leading cause of morbidity and mortality among men2,3,4). Therefore, Development of novel therapeutic agents or approaches is urgent to overcome CaP.
Apoptosis is a mechanism by which cells execute an endogenous programmed cell deaths in response to various stimuli, including chemotherapeutic agents, ionizing radiation, tumor necrosis factor α, fas ligand, dexamethasone, and other agents of stress or injury. It is mediated through multiple pathways, involving a complex array of biochemical regulators and molecular interactions. The well defined set of biochemical and morphological changes that characterize apoptosis include caspase activation, release of cytochrome c, nuclear condensation, protease activation, and DNA fragmentation5).
As apoptosis has become a potential therapeutic target for the inhibition of cancer growth recently6,7), understanding of the molecular mechanisms of apoptosis is regarded to be crucial for selective therapy.
Previous studies demonstrated that even an androgen independent prostate carcinoma cells had a tendency to be responsive to specific agents, undergoing growth arrest and apoptosis8,9). This has profound therapeutic implications for the potential treatment of androgen resistant advanced prostate cancer10).
Lee et al11) revealed that bee venom (BV) can inactivate NF-κB and suppress the expression of inflammation related proteins such as iNOS, COX- 2 and cPLA2 , and thereby prevent anti-apoptotic ability of NF-κB causing human hormone dependent LNCaP cells undergo apoptosis.
Meanwhile, the growth of prostate cancer are dependent on androgens, which act through the action of androgen receptor (AR)12). Therefore, androgen deprivation therapy (ADT) consisting of reduction in the availability of androgens and/or interference with their function via the AR, remains the most important therapy for advanced Prostate Cancer13). However, as the disease progresses, ADT eventually fails leading to the transition of androgen dependent prostate cancer to androgen independent prostate cancer14). In order to better understand the role of the AR in prostate cancer development and progression, it is important to elucidate the AR signaling cascades and the genes that are subject to AR control, because AR and its signaling pathways are strongly associated with the development and progression of prostate cancer, even in androgen independent tumors.
According to a previous report15), inhibition of Akt, COX-2, and MMP-9 in these pathways may exhibit profound effects on prostate tumor growth and metastasis, suggesting its potential to be available for treating both androgen dependent and androgen independent prostate cancer, and to exert synergic effect on delaying the onset of androgenindependent prostate cancer in combination with ADT.
In present study, to confirm the effect of BV and melittin (MT) on androgen dependent prostate cancer growth, I therefore investigated whether their inducing AR related apoptosis through inhibition of Akt, COX-2, and MMP-9 in AR and its signaling pathways in vitro, and On the basis of the previous Lee's finding and in vitro finding of this study, I also investigated whether their exerting influenced upon genes in AR signaling pathway and apoptosis regulatory proteins and their protease substrates in LNCaP xenograft nude mice in vivo.
Dried bee venom (BV) was purchased from You- Miel Bee Venom Ltd. (Hwasoon, Jeonnam, Korea). The composition of the BV was as follow: 45~50% melittin, 2.5~3% apamin, 2~3% MCD peptide, 12% PLA2, 1% lyso-PLA, 1~1.5% histidine, 4~5% 6pp lipids, 0.5% secarpin, 0.1% tertiapin, 0.1% procamine, 1.5~2% hyaluronidase, 2~3% amine, 4~5% carbohydrate, and 19~27% other, including protease inhibitor, glucosidase, invertase, acid phosphomonoesterase, dopamine, norepinephirne, and unknown amino acids, with >99.5% purity. Melittin (MT) was purchased from Sigma Chemical Co. (St. Louis, MO, USA). goat polyclonal antibody to COX-2 (1:500), and all of the secondary antibodies such as Akt, p-Akt, Bax, Bcl-2, p53, PARP, caspase- 3, -9, MMP-9, calpain used in Western blot analysis were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). T4 polynucleotide kinase was obtained from Promega (Madison, WI). Poly (dI-dC), horseradish peroxidase- labeled donkey anti-rabbit secondary antibody, and ECL detection reagent were obtained from Amersham Pharmacia Biotech (Piscataway, NJ). Reagents for sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis were purchased from Bio-Rad (Hercules, CA). All other reagents were purchased from Sigma unless otherwise stated.
The LNCaP human prostate cancer cell was obtained from ATCC (American Type Culture Collection, Rockville, MD). Prostate cells were cultured in RPMI-1640 medium (Life Technologies Inc., Gaithersberg, MD) supplement with 10% fetal calf serum (FCS; Collaborative Biomedical Products,Bedford,MA)and antibiotics, penicillin/streptomycin( 100unit/mL, Bioproducts, Walkersville, MD) Cell cultures were then maintained at 37℃ in a humidified atmosphere of 5% CO2.
Apoptosis assays were performed using the 4,6- diamidino-2-phenylindole (DAPI) staining. LNCaP cells were cultured in the absence or presence of increasing concentrations of melittin or bee venom, and apoptosis induction were evaluated after 24 hrs. Apoptotic cells were determined by the morphological changes after DAPI staining under fluorescence microscopic observation (DAS microscope, 100 or 200x; Leica Microsystems, Inc., Deerfield, IL). For each determination, three separate 100-cell counts were scored. Apoptosis was expressed as a percentage calculated from the number of cells with apoptotic nuclear morphology divided by the total number of cells counted.
Apoptosis was also evaluated by TUNEL staining assay. In short, cells were cultured on 8-chamber slides. After treatment with melittin (0.5-2.5 ㎍) or bee venom (1-10 ㎍) for 24 hrs, the cells were washed twice with PBS and fixed by incubation in 4% paraformaldehyde in PBS for 1hr at room temperature. TUNEL assays were performed by using the in situ Cell Death Detection Kit (Roche Diagonostics GmbH, Mannheim, Germany) according to manufacturer’s instructions. Total number of cells in a given area was determined by using DAPI nuclear staining. The apoptotic index was determined as the number of TUNEL-positive stained cells divided by the total cell number counted 100.
4. Anti-tumor activity of bee venom in vivo xenograft
Six-week old male BALB/c athymic nude mice were purchased from Japan SLC (Hamamatsu, Japan). Prostate cancer LNCaP cells were injected subcutaneously (1x107 tumor cells/0.1 mL PBS/animals) with a 27-gauge needle into the right lower flanks of the mice. Tumors were removed from mice, and same size tumor fragments (3×3×3㎜) were then implanted s.c. into the right flank of other BALB/c athymic nude mice with a 13-gauge trocar needle. When the tumors had reached an average volume of 100-300 mm3 at 14 day, the tumor-bearing nude mice were intraperitoneally injected with bee venom (3 and 6 mg/kg, twice per week for 4 weeks). The group treated with saline was used as the control and the group treated with cisplatin (5 mg/kg, five times, every five days for 4 weeks) as positive control. The weight and tumor volume of the animals were monitored once per week. The tumor volumes were measured with vernier calipers and calculated by the following formula: (A × B2)/2, where A is the larger and B is the smaller of the 2 dimensions of the tumor. Relative tumor volume (100%) was calculated by actual tumor weight (ATW) over initial tumor weight (ITW, day 0, control), as follows: ATW÷ITW×100%. Measurements were taken every three or four days.
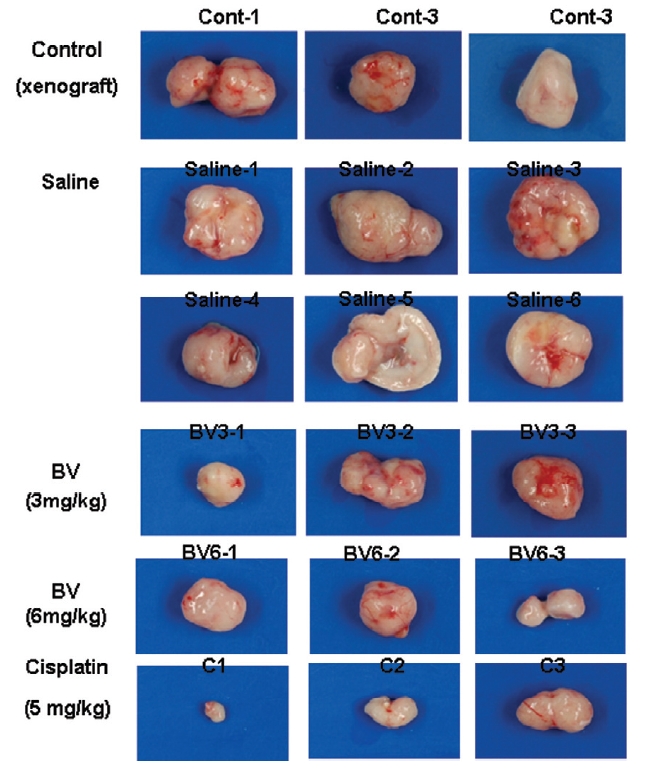
At the end of the experiment, the animals were sacrificed with cervical dislocation. The tumors were separated from the surrounding muscles and dermis, excised and weighed.
Combination of the three randomly selected tumor tissues each from ten individual mouse in control, positive control and bee venom-treated groups were used for total cell lysate preparation and analyzed by western blot analysis, EMSA and Apoptosis evaluation.
LNCaP cells or Tumor fragments were homogenized with lysis buffer [50 mM Tris pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.2% SDS, 1 mM PMFS, 10 μl/mL aprotinin, 1% igapel 630 (Sigma- Aldrich, St. Louis, MO, USA), 10 mM NaF, 0.5 mM EDTA, 0.1 mM EGTA and 0.5% sodium deoxycholate], and centrifuged at 23,000 g for 1 hr. Equal amount of proteins (80 μg) were separated on a SDS/12%-polyacrylamide gel, and then transferred to a nitrocellulose membrane (Hybond ECL, Amersham Pharmacia Biotech Inc., Piscataway, NJ). Blots were blocked for 2 hrs at room temperature with 5% (w/v) non-fat dried milk in Trisbuffered saline [10 mM Tris (pH 8.0) and 150 mM NaCl] solution containing 0.05% tween-20. The membrane was incubated for 5 hrs at room temperature with specific antibodies rabbit polyclonal for Akt, p-Akt, p53, Bax (1:500 dilution, Santa Cruz Biotechnology Inc. Santa Cruz, CA, USA), Bcl-2, PARP, cleaved caspase-3, -9, calpain and MMP-9 (1:1000 dilution, Cell Signaling Technicology, Inc. Beverly, MA, USA), goat polyclonal antibody to COX-2 (1:500). The blot was then incubated with the corresponding conjugated anti-rabbit immunoglobulin G-horseradish peroxidase (Santa Cruz Biotechnology Inc.). Immunoreactive proteins were detected with the ECL western blotting detection system. The relative density of the protein bands was scanned by densitometry using MyImage (SLB, Seoul, Korea), and quantified by Labworks 4.0 software (UVP Inc., Upland, California).
6. Preparation of nuclear extracts and electromobility shift assays
It was performed according to the manufacturer's recommendations (Promega, Madison, WI). Briefly, 1 × 106 cells/mL from the tumor fragments of each group was washed twice with 1× PBS, followed by the addition of 1 mL of PBS, and the cells were scraped into a cold Eppendorf tube. Cells were spun down at 15,000 g for 1 min, and the resulting supernatant was removed. Solution A (50 mM HEPES, pH 7.4, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 0.1 μg/mL phenylmethyl- sulfonyl fluoride, 1 μg/mL pepstatin A, 1 μg/mL leupeptin, 10 μg/mL soybean trypsin inhibitor, 10 μ g/mL aprotinin, and 0.5% Nonidet P-40) was added to the pellet in a 2:1 ratio (v/v) and allowed to incubate on ice for 10 min. Solution C (solution A + 10% glycerol and 400 mM KCl) was added to the pellet in a 2:1 ratio (v/v) and vortexed on ice for 20 min. The cells were centrifuged at 15,000 g for 7 min, and the resulting nuclear extract supernatant was collected in a chilled Eppendorf tube. Consensus oligonucleotides were end-labeled using T4 polynucleotide kinase and [g-32P] ATP for 10 min at 37℃. Gel shift reactions were assembled and allowed to incubate at room temperature for 10 min followed by the addition of 1 μl (50,000- 200,000 cpm) of 32P-labeled oligonucleotide and another 20 min of incubation at room temperature. For superscript assays, nuclear extracts from the cells of combination of the three randomly selected tumor tissues each from ten individual mouse in control, cisplatin (5 mg/kg) treated positive control and bee venom (3 mg/kg or 6 mg/kg)-treated groups were incubated with specific antibodies against the Rel-A NF-κB isoforms for 1 hr before EMSA. For competition assays, the nuclear extracts were incubated with unlabelled NF-κB oligonucleotide (50X, 100X and 200X) or labeled SP-1 (100X) and AP-1 (100X) for 30 min before EMSA. Subsequently 1 μl of gel loading buffer was added to each reaction and loaded onto a 6% nondenaturing gel and electrophoresed until the dye was threefourths of the way down the gel. The gel was dried at 80℃ for 1 hr and exposed to film overnight at 70 ℃. The relative density of the DNA-protein binding bands was scanned by densitometry using MyImage (SLB, Seoul, Korea), and quantified by Labworks 4.0 software (UVP Inc., Upland, California).
Data were analyzed using one-way analysis of variance followed by Tuckey test as a post hoc test. Differences were considered significant at p<0.05.
To delineate whether the inhibition of LNCaP cell proliferation by the MT or BV was due to increase of the apoptosis induction, I evaluated change of the chromatin morphology of human prostate cancer cells using DAPI staining. Consistent with the decrease of the cell viability, apoptosis determined after 24 hrs treatment was increased in a dose dependent manner. The percentages of the control significantly increased by MT 2.5 μg/mL was 75.8±14.2%, which of the control significantly increased by BV 5 and 10 μg/mL was 33.3±13.1 and 42.9±10.0% respectively (Table 1, Fig. 1). I also evaluated LNCaP cell apoptosis by TUNEL assay. As shown in Fig. 1 TUNEL-positive cells (stained green) were dose-dependently increased in MT or BV treated LNCaP cells, and the nuclei (stained blue) were found to be condensed (Fig. 1).
2. Expression of apoptosis and androgen receptor regulatory proteins
Androgen Receptor (AR) plays an important role in the initiation and progression of prostate cancer by regulating cell proliferation, differentiation and apoptosis16).
AR signaling regulates the Akt, COX-2, MMP-9 activity. The Akt signaling also regulates AR phosphorylation and its activity. PSA, prostate-specific antigen.
Considering of the correlation of androgen deprivation therapy, and Akt, COX-2 and MMP-9 activity, androgen-dependent prostate cancer progression is controlled by reducing AR activity with androgen deprivation therapy. Concomitantly, androgen deprivation therapy may enhance activities of Akt, COX-2 and MMP-9, which in turn promotes the androgen-independent transition15).
Calpain in prostate cancer cells is also activated upon induction of apoptosis by chemotherapeutic agents, and activated calpain is one of the proteases responsible for AR degradation17).
COX-2, Akt, MMP-9 involve androgen receptor breakdown and apoptosis of androgen dependent prostate cancer as down regulators, dissimilar with calpain, pro-apoptotic protein and AR degradation promotors, implying their involving the transition of androgen dependent prostate cancer to androgen refractory one.
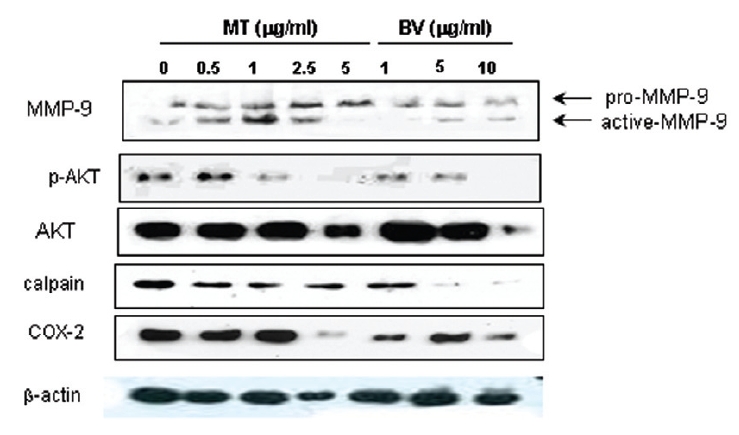
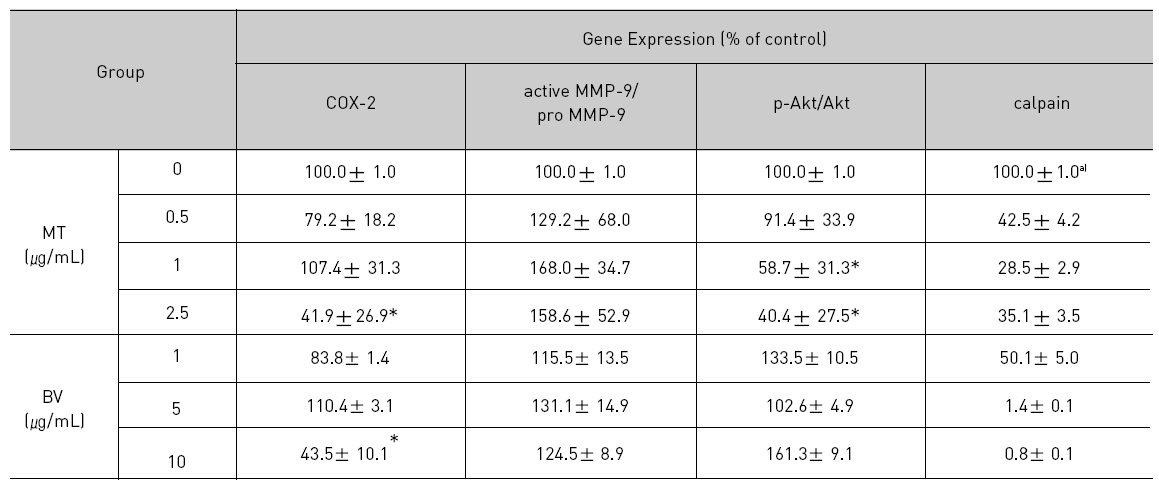
Therefore, to assess whether MT and BV exerts effect on proliferation of LNCaP cells via apoptosis with AR related proteins in the center, a western blot analysis of COX-2, Akt, MMP-9 and calpain was performed, and the increase of apoptotic action was generally confirmed by the ability of MT or BV to induce down regulation of anti-apoptotic and AR regulatory COX-2, Akt, MMP-9, calpain based upon their expressions of Fig. 2. in LNCaP cells treated with a different dose of MT or BV. Compared with control, Expression of COX-2 significantly decreased by 2.5 μg/mL of MT or 10 μ g/mL of BV was 41.9±26.9 or 43.5±10.1%. p- Akt/Akt was used as a measure of activation of Ak which significantly decreased by 1 and 2.5 μg/mL of MT was 58.7±31.3 and 40.4±27.5%(Table Ⅱ, Fig. 2). On the contrary, Akt activity in BV, and MMP-9 activity measured by active MMP-9/pro- MMP-9 in both BV and MT did not show decrease of their expressions with having a tendency to increased and calpain was decreased in the BV and MT as well (Table Ⅱ, Fig. 2).
3. Effect of BV on tumorigenesis of LNCaP prostate cancer cells in an athymic nude mouse model
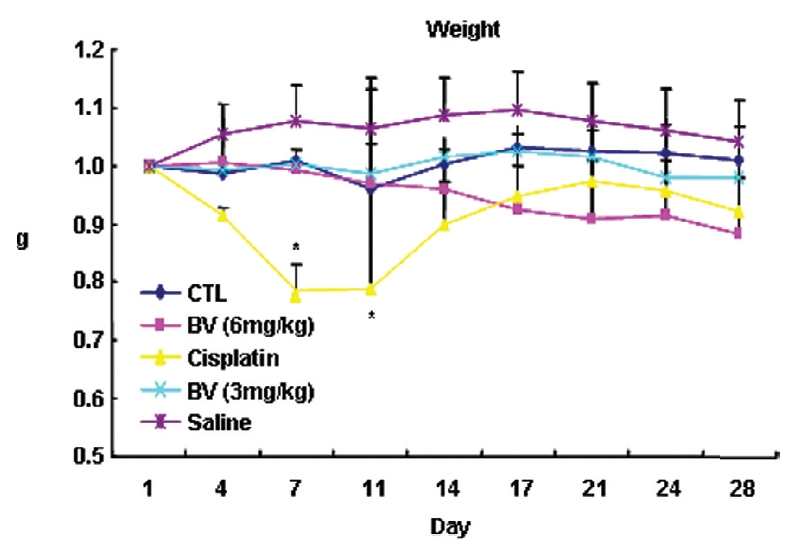
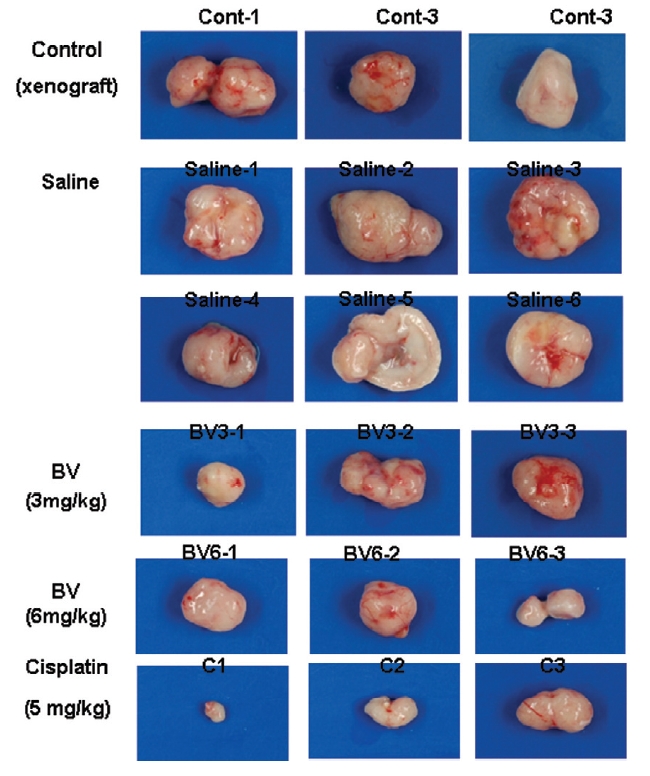
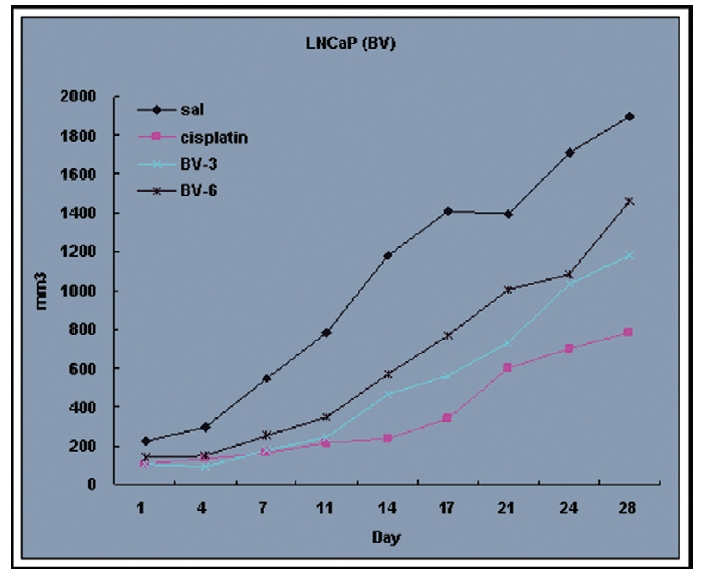
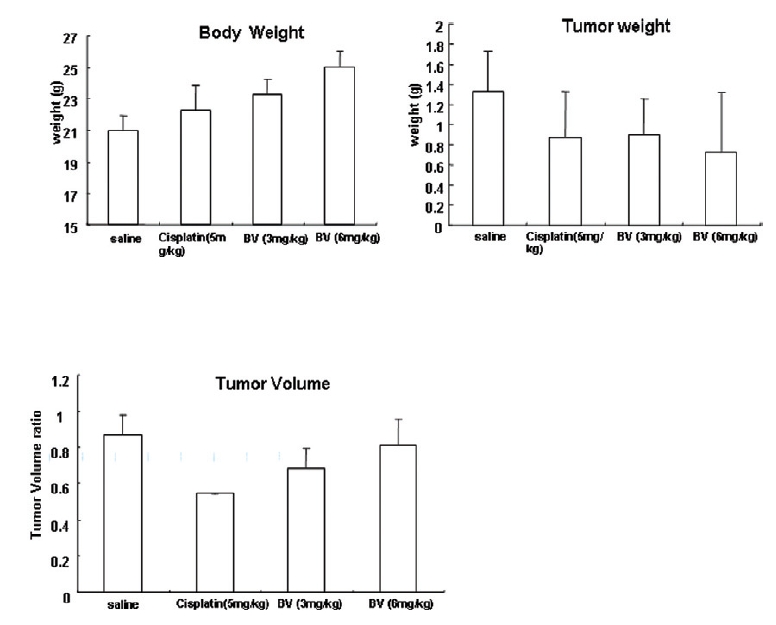
Following the above findings MT or BV induce apoptosis via down regulation of AR and apoptosis related proteins in the LNCaP human hormone sensitive prostate cancer cells in vitro, To elucidate the anti-cancer effect of BV in vivo, I investigated the tumor growth induced by apoptosis in prostate xenograft-bearing nude mice. BV did not cause any loss in the body weight (Fig. 6), and affect food intake suggesting that BV did not exhibit apparent signs of toxicity in animals. Tumor volume and tumor weight were measured weekly, and all mice were sacrificed at the end of 4 week, when tumors were dissected and measured (Fig. 3).
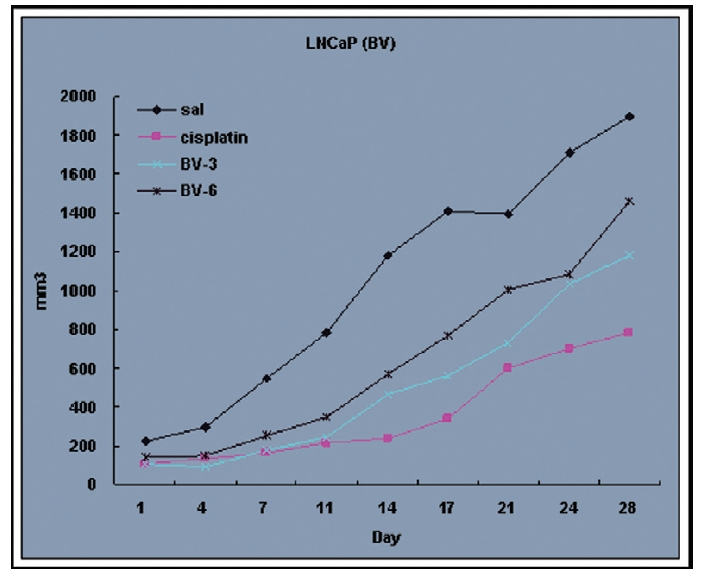
Fig. 4 and 5 represent tumor volume and weight, respectively, measured in each groups treated by BV (3 and 6 mg/kg, n=10) or cisplatin (5 mg/kg, 5 times, n=5) or saline (n=10), suggesting Tumor growth was gradually and time-dependently retarded by the treatment of BV similarly with cisplatin, compared with sham group.
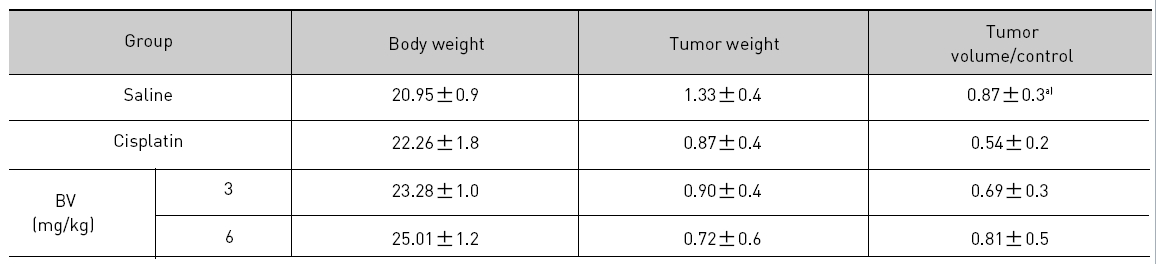
At the final, Tumor volumes ratio in 3 or 6 mg/kg BV-treated mice (0.69±0.3 or 0.81±0.5) were smaller than those of sham group (0.87±0.3). Tumor weights in 3 or 6 mg/kg BV-treated group (0.90±0.4 or 0.72±0.6) were also lighter than sham group (1.33±0.4) (Fig.6, TableⅢ).
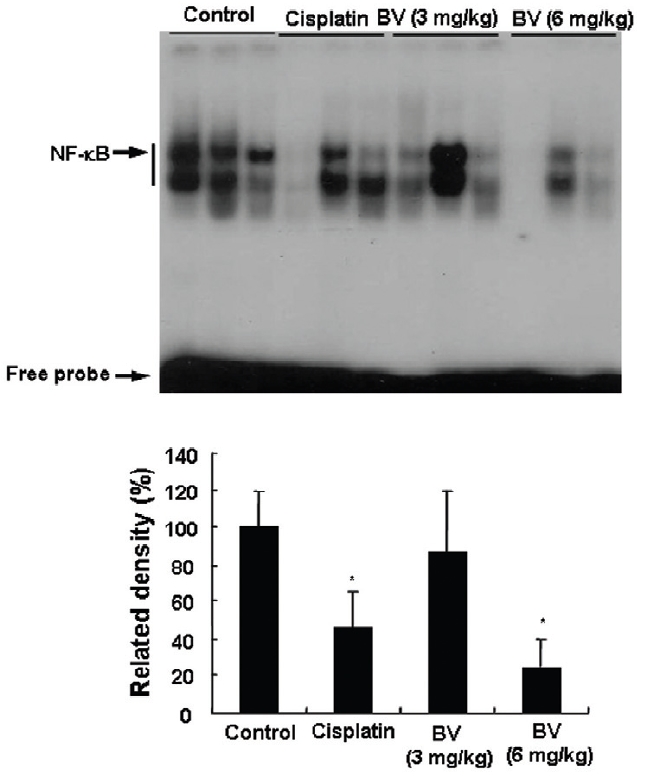
4. Inhibition of NF-κB in LNCaP Xenografts Nude Mice
NF-κB is known to inhibitory transcription factor of apoptosis. According to Lee et al’s report11), BV can inactivate NF-κB and suppress the expression of inflammation related genes such as iNOS, COX-2 and cPLA2 , and thereby prevent anti-apoptotic ability of NF-κB causing LNCaP cells go apoptosis. To investigate hypothesis whether this findings also works in vivo, I assessed NF-κB activity in LNCaP xenografts nude mice. Following animals sacrificed at the final, and the tumors from three randomly selected mice were separated from the surrounding muscles and dermis, 1× 106 cells/mL from the tumor fragments of each group was washed twice with 1× PBS after the addition of 1 mL of PBS, and EMSA was performed as described in the material and method. NF-κB was highly activated in the xenografts (controls), however, the activation of NF-κB were gradually decreased by 3 mg/kg or 6 mg/kg of BV in the BV treated groups as well as in the cisplatin positive control groups (Fig.7). The density of control significantly decreased by 6 mg/kg of BV was 22.3±23.3, even lower than 42.4 ±21.4 of cisplatin (Table Ⅳ).
5. Expressions of apoptosis regulatory proteins in LNCaP xenografts
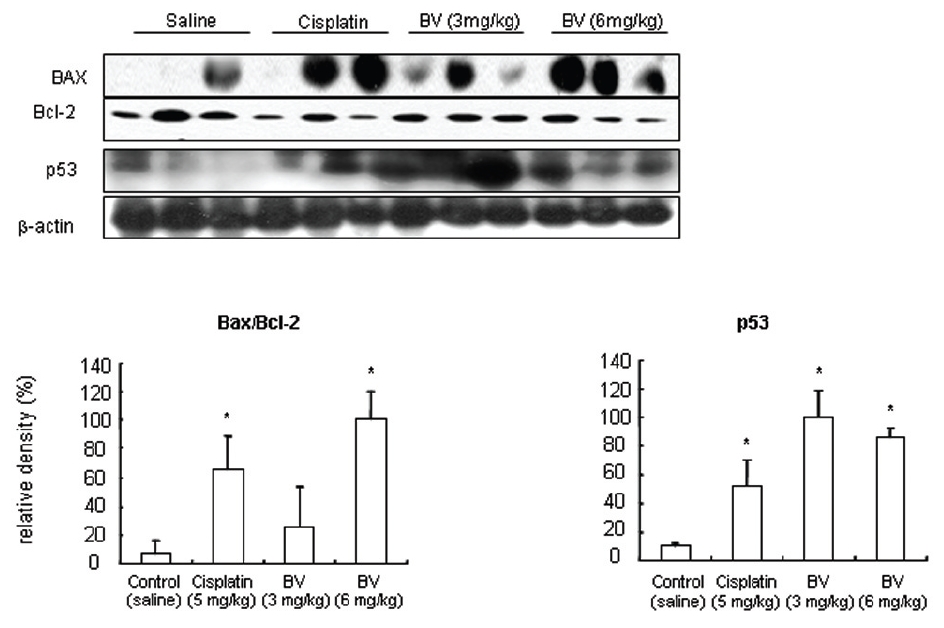
p53 is known as a tumor suppressor gene and a transcriptional factor, which plays a major role in apoptosis. By stimulating Bax, one of p53 target genes and down regulating anti-apoptotic protein such as Bcl-2. Dimerizling with Bcl-2, Bax generally predominate over it, resulting in inhibition of cancer growth through vivid apoptosis via activating caspase family such as caspase-3 and caspase-9. AR in hormone dependent prostate cancer is a potential target for androgen deprivation therapy, which is affected by apoptosis regulatory proteins such as COX-2, MMP-9, calpain and Akt. The present in vitro study revealed MT or BV induced apoptosis through possibly exerting inhibitory effect on AR via down regulation of anti-apoptotic and AR regulatory COX-2, Akt, MMP-9, calpain in LNCaP prostate cancer cells. To reconfirm these in vitro findings in the xenografts in vivo, and to elaborate how BV inhibit prostate cancer growth by inducing apoptosis, I investigated the effects of BV on apoptosis regulatory genes including Bax, Bcl-2, p53, COX-2, MMP-9, calpain, caspase-3 and caspase- 9 in LNCaP xenograft nude mice. The tumor fragments from randomly selected mice of each group were homogenized in lysis buffer Pro-Prep, and then, equal amounts of proteins (80μg) were studied under the same conditions and course as in vitro.
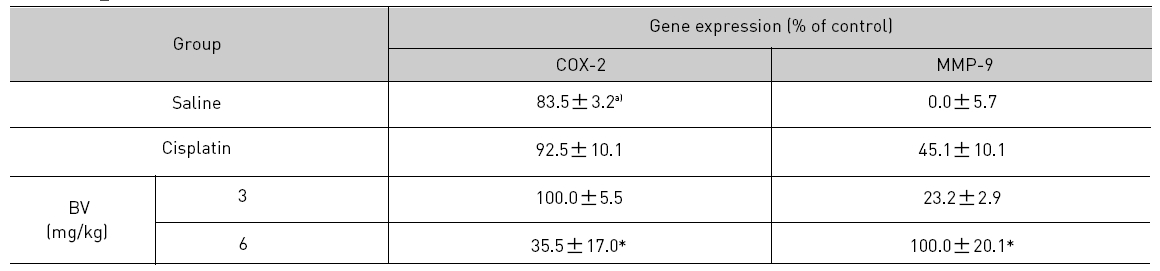
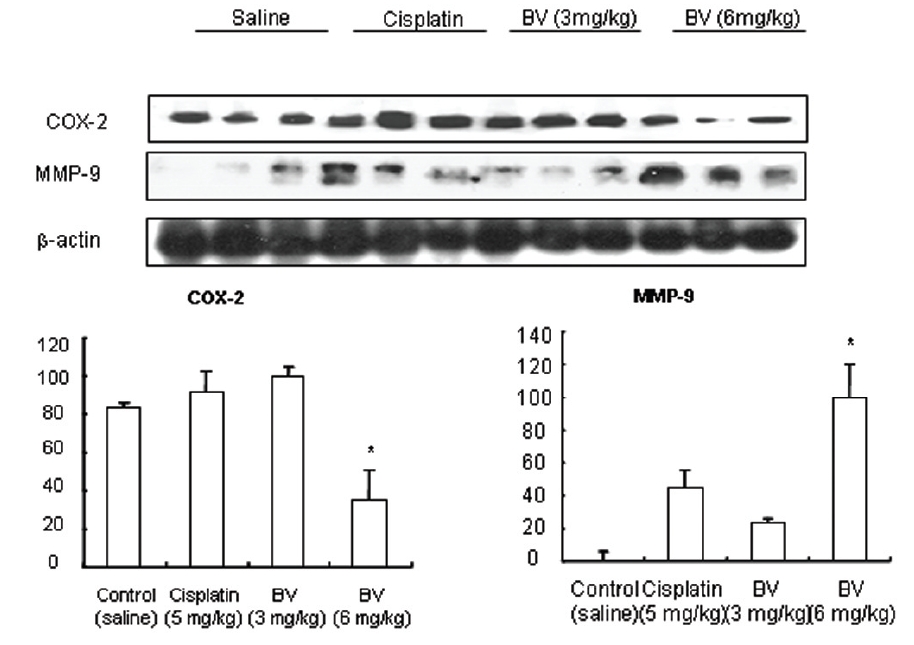
As in the cisplatin group, p53 activated one of its target gene, pro-apoptotic Bax, subsequently predominating over anti-apoptotic Bcl-2 in 3 or 6 mg/kg of BV-treated groups, compared with sham control (Fig.8). p53 significantly increased by 3 or 6 mg/kg of BV was 100.0±18.5 or 85.3±6.5, and Bax/Bcl-2 ratio significantly increased by 6 mg/kg of BV was 100.0±20.1 (TableⅤ). By vivid apoptosis going, caspase-3 and -9 were activated consecutively (Fig.9). Caspase-3 and caspase-9 significantly increased dose-dependently by 3 or 6 mg/kg of BV was 79.5±15.2 or 100.0±12.3 and 75.6± 3.4 or 100.0±10.5 respectively (TableⅥ). Calpain was also activated during apoptosis, contributing to AR break down, which significantly increased by 6 mg/kg of BV was 100.0±18.6 (Fig. 9, TableⅥ). COX-2 and MMP-9 were regarded as anti-apoptotic proteins, but activity of MMP-9 was unexpectedly increased under BV inducing apoptosis in LNCaP xenografts, although that of COX-2 was significantly decreased to 35.5±17.0 by 6 mg/kg of BV and MMP-9 was significantly increased to 100.0±20.1 by 6mg/kg of BV (Fig.10, TableⅦ).
The central and noteworthy findings in the present study is the identification of anticancer efficacy of MT or BV on human androgen sensitive prostate carcinoma LNCaP xenografts in vivo as well as the cells in vitro. Most of the present available cytotoxic anticancer drugs exert their effect through induction of apoptosis in cancer cells18,19), which is regarded as one of the major mechanisms for the targeted therapy of various cancers18-21).
Prostate cancer has the highest incidence among malignancies in males, and is the second leading cause of cancer-related death in Western countries22).
Early stages of prostate cancer can be effectively treated by androgen- deprivation therapy through surgical and medical castration. However, most of these prostate cancer patients eventually relapse to a hormone-refractory state that no longer responds to androgen ablation23).
AR plays an important role in the initiation and progression of prostate cancer by regulating cell proliferation, differentiation and apoptosis24),which also appears to be a dominant factor in the transition from hormone-sensitive to hormone- refractory disease, considering well-established evidence to show that the AR gene undergoes alterations such as amplification or mutation in hormone independent cancers25). Therefore, novel strategies and agents are needed to be developed for delaying the transition, keeping up androgen dependent curable state of prostate cancer. According to review of Chang et al15), Expression of Akt, COX-2, and MMP-9 is associated with tumor progression through affecting AR and AR signal pathway, and androgen deprivation and other anti-AR therapies carry the seeds of their own failure by up regulating Akt, COX-2, and MMP-9. Thus, inhibition of the Akt, COX-2, and MMP-9 pathways could be an effective therapeutic approach for advanced prostate cancer. Moreover, the androgen independence state could be delayed by combining androgen deprivation therapy with inhibition of the Akt, COX-2, and MMP-9 pathways.
As another class of cysteine proteases and calcium dependent proteases, calpains, may play a role in apoptosis and degrade a number of important cellular proteins including protooncoproteins, steroid hormone receptors, protein kinases, and cytoskeleton proteins, implying activated calpain is one of the proteases responsible for AR degradation26) and concomitant apoptosis in the advanced prostate cancer.
In the present study, I evaluated apoptosis of LNCaP cells by TUNEL assay. TUNEL-positive cells (stained green) were dose-dependently increased in MT or BV treated LNCaP cells, and the nuclei (stained blue) were found to be condensed, suggesting BV or MT induced apoptosis go vividly, and I also investigated the Effect of MT or BV on activities of Akt, COX-2, MMP-9 and calpain by western blot analysis in the human androgen refractory LNCaP prostate cancer cells, in vitro in order to confirm the mentioned above. My data demonstrated BV or MT induced inhibition of LNCaP cell proliferation through AR related apoptosis via down regulation of activities of Akt, COX- 2, MMP-9, though calpain was actually down regulated and revealed possible relation with the transition of androgen dependent state into androgen refractory state. COX-2 was also inactivated by BV, reconfirming the above in vitro findings, and calpain and MMP-9 was up-regulated contrarily to them in the xenografts. Thus, down regulation of COX-2, Akt or MMP-9 and up-regulation of calpain suggested that MT or BV affect AR and its signal pathway such as AR degradation and could play a major role in repressing the transition of androgen dependent state to androgen refractory one in LNCaP prostate cancer cells and the xenografts as well.
Apoptosis, a programmed cell death triggered by various stimuli, is characterized by a series of distinct biochemical and morphological changes27), including increase in ROS level28), activation of caspases, cell shrinkage, chromatin condensation and nucleosomal degradation29). It plays a crucial role in eliminating the mutated hyperproliferating cells from the system, although it happens in pathological as well as physiological conditions. Therefore, induction of apoptosis in tumor cells may be considered as a protective mechanism against development and progression of cancer, which was related with a variety of therapeutic methods, including irradiation, cytotoxic chemotherapy, and hormone manipulation30). A number of genes are involved in the apoptosis control, including the protooncogene Bcl-231,32) and the tumor suppressor gene p5333). Bcl- 2 was initially identified in B-cell leukemia and non-Hodgkin's lymphoma. The Bcl-2 gene product down regulates apoptosis, but actually has no direct effect on cell growth34), and is expressed in the cytoplasm of basal epithelial cells in the normal and hyperplastic prostate35). A family of genes with sequence homology to Bcl-2 has been identified, forming two functionally antagonistic groups which control the balance between cell death and survival. Bcl-2 is a cell death suppressor, whereas Bax is a cell death promoter32,34-36). Under apoptosis, Bax generally overwhelms Bcl-2. Previous studies investigating Bcl-2 in prostate cancer showed that overexpression of Bcl-2 protein is associated with increasing tumor stage37) and the development of hormone-refractory disease38-40), to which, presently, inactivation of the tumor suppressor gene p53 through mutation commonly identified in human cancers is also related32,41). The combination of p53 and Bcl-2 overexpression in prostate cancer has been shown to be a good predictor of postradical prostatectomy recurrence27,28,42,43), and correlates with hormone-refractory prostate cancer40).
Another significant event in apoptosis is mitochondria dysfunction. Loss of mitochondria transmembrane potential (MTP) elicits the release of cytochrome c from mitochondria to cytosol44). After release, it promotes caspase-9 and -3 subsequently activating downstream caspase-345).
To elaborate how BV inhibit prostate cancer growth by inducing apoptosis, I investigated the effects of BV on apoptosis regulatory genes including Bax, Bcl-2, p53, caspase-3 and caspase-9 in LNCaP xenograft nude mice, additionally to in vitro.
As a result, It was demonstrated that compared with the cisplatin group, BV activated p53, up-regulating one of its target gene, pro-apoptotic Bax, and subsequently predominating over anti-apoptotic Bcl-2, and finally undergoing apoptosis vividly through promoting caspase-3 and -9 in this study. These findings implied that BV could also be adopted for a useful agent to inhibit prostate cancer through apoptosis induction and delay of transition to hormone refractory state.
The rate of cell proliferation is low in prostate cancer and is a predictive factor for survival29,46).
I investigated the tumor growth in prostate xenograft-bearing nude mice to elucidate the antiproliferative effect of BV on LNCaP cells in vivo. BV did not cause any loss in the body weight, and change in the food intake implying that BV did not exhibit toxic effect on the animals. Tumor growth estimated by tumor volume and tumor weight was gradually and time-dependently retarded by BV, reconfirming anti- proliferative effect of BV, in vitro and suggesting BV could affect survival of advanced prostate cancer.
It has been well established that NF-
Based on a previous reports11,51) BV played a crucial role in inhibiting growth of prostate cancer through induction of apoptosis via inactivation of NF-
[Table 1.] Effect of BV on apoptosis in LNCaP Cells
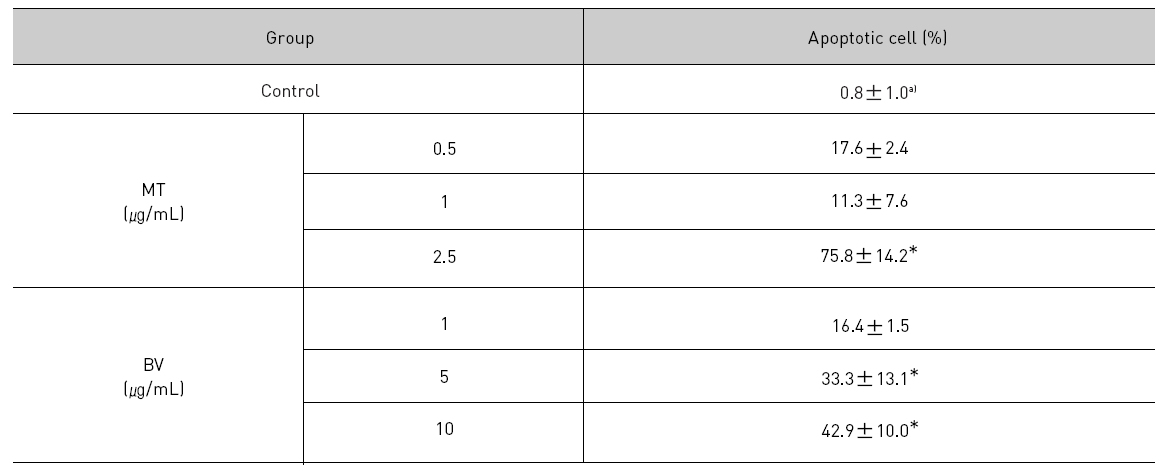
Effect of BV on apoptosis in LNCaP Cells
[Table 2.] Effect of BV on AR related apoptosis in LNCaP Cells
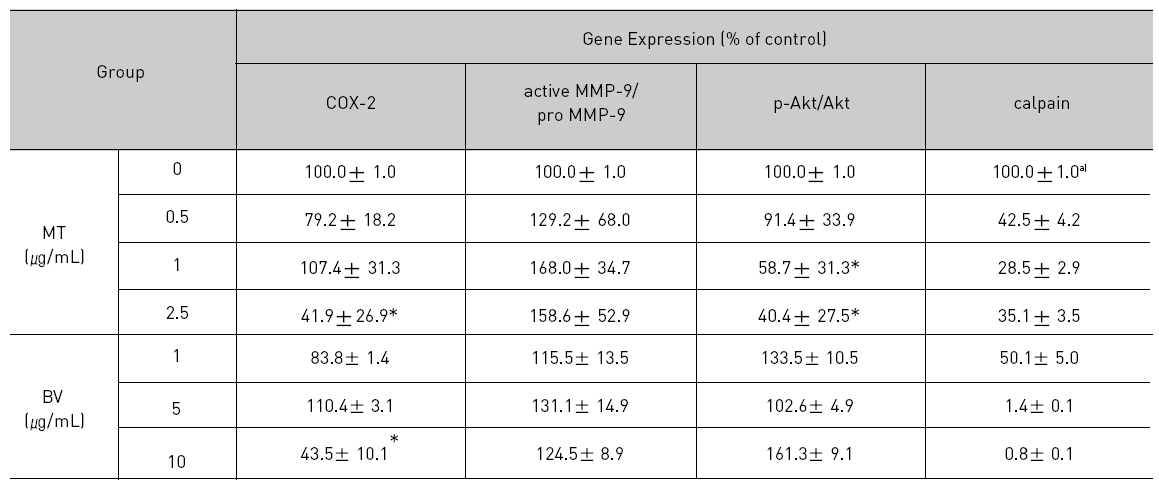
Effect of BV on AR related apoptosis in LNCaP Cells
[Table 3.] Effect of BV on body weight tumor weight and tumor volume ratio at the final day
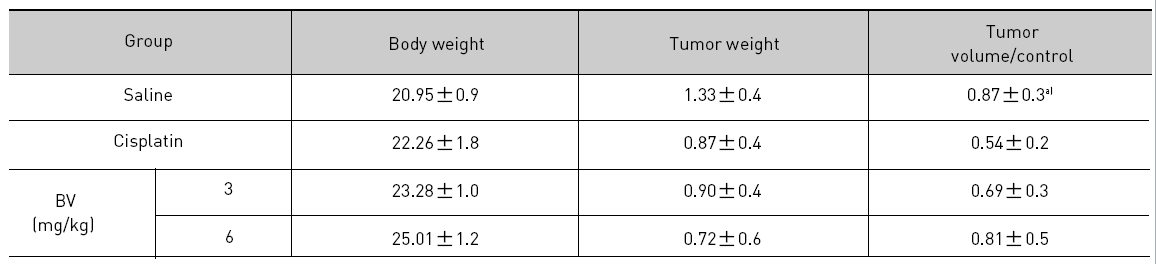
Effect of BV on body weight tumor weight and tumor volume ratio at the final day
[Table 4.] Effect of BV on NF-κB activity in xenograft nude mice
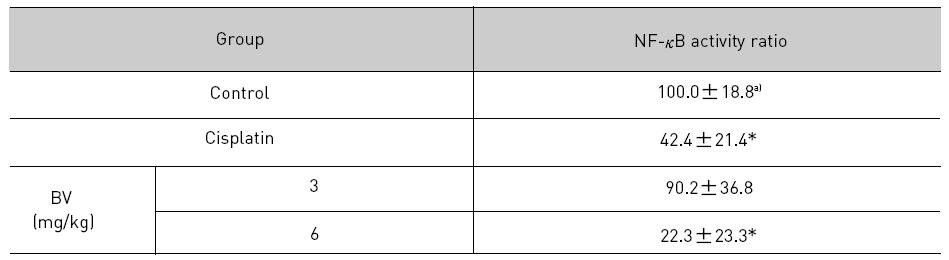
Effect of BV on NF-κB activity in xenograft nude mice
[Table 5.] Effect of BV on major apoptosis regulatory genes in LNCaP xenografts
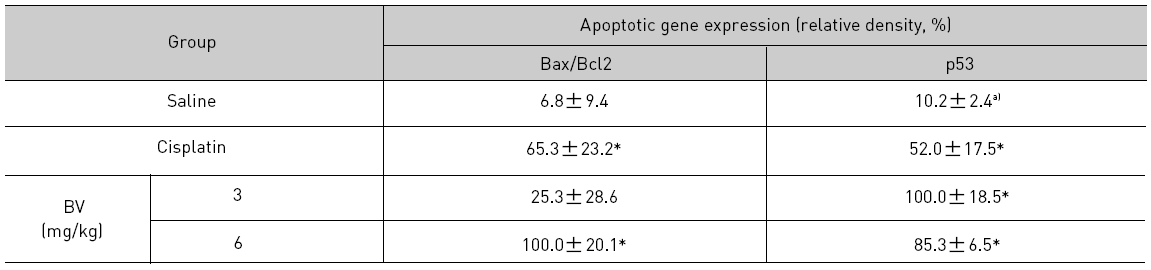
Effect of BV on major apoptosis regulatory genes in LNCaP xenografts
[Table 6.] Effect of BV on apoptosis related cysteine proteases in LNCaP xenografts
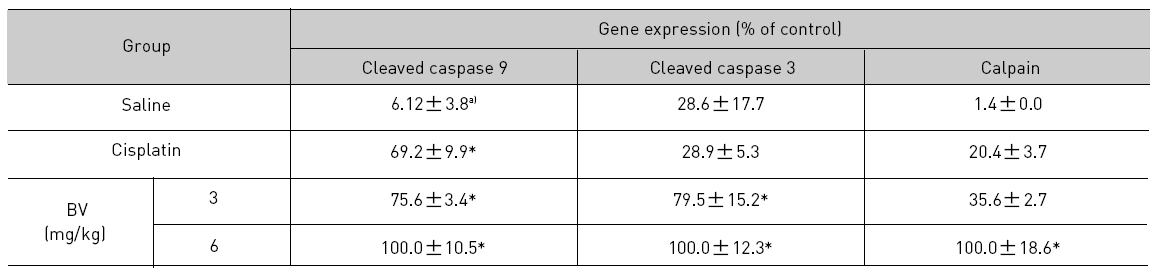
Effect of BV on apoptosis related cysteine proteases in LNCaP xenografts
[Table 7.] Effect of BV on anti-apoptotic genes in LNCaP xenografts
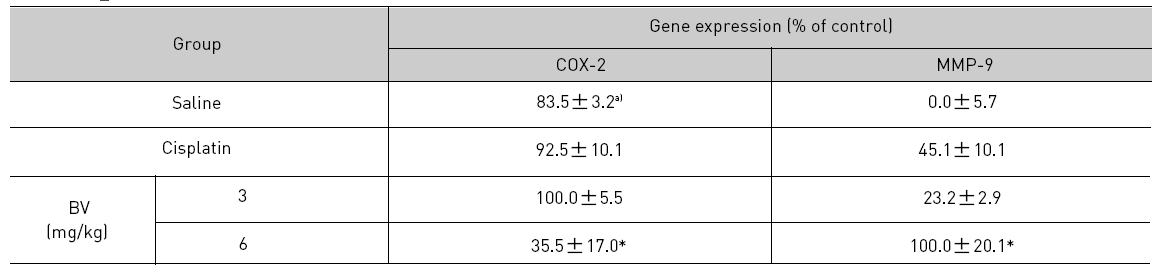
Effect of BV on anti-apoptotic genes in LNCaP xenografts