In this study, a computational chemistry methodology called as molecular modeling was been applied to explain several experiment results mechanistically. The reaction chosen for this study was to remove carbon dioxide, known as a primary greenhouse gas, by an epoxide via the carbon dioxide fixation to produce carbonates. This reaction inherently needs the use of catalysts because it has a significantly high activation barrier (55~59 kcal/mol). Among various types of catalysts, we studied in zeolitic imidazolate framework 90 (ZIF-90)/ionic liquid immobilized ZIF-90 (IL-ZIF-90), polystyrene-supported quaternized ammonium salt, KI/KI-glycine, and dimethylethanolamine (DMEA). First, probable reaction pathways were proposed based on calculated energetics by computational chemistry. The energetics was then used for the thermodynamic interpretation on the activity of catalysts. In the case of ZIF-90/IL-ZIF-90 and KI/KI-glycine, IL-ZIF-90 and KI-glycine showed better yields compared to their counterparts. The calculation proposed interesting results that it is not from the lowering of activation energy but from the unstable intermediates of ZIF-90 and KI-glycine. For DMEA, the calculated activation energy was ~42 kcal/mol, much lower than that of the non-catalytic reaction. A possible reaction pathway was located to confirm the interaction between −NH group from ammonium and oxygen from epoxide for polystyrene-supported quaternized ammonium salt.
화합물의 구조에 대한 이해는 물리, 화학, 신소재, 화공 분야에서 매우 중요한 연구 분야로 꾸준히 관심의 대상이 되고 있다. 최근에 컴퓨터를 사용하여 화합물의 구조를 파악하고 에너지를 계산하는 계산화학이 다각적으로 이용되고 있는데[1], 이는 실험을 배제하고 물리화학 이론들과 컴퓨터의 계산 능력만을 이용하여 연구를 수행하는 방법을 말한다. 특히 계산화학은 실험이나 분석에 의해 밝히기 어려운 반응의 메카니즘을 이론적으로 규명함으로써 실험 결과를 뒷받침하는데 유용하게 활용될 수 있다.
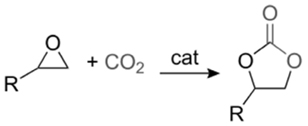
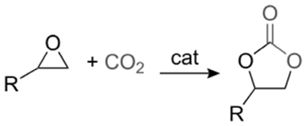
산업혁명 이후, 중화학공업의 성장으로 다양한 오염물질이 대기 중으로 배출되어 온실효과를 일으킴으로써 지구 온난화 현상을 발생시켜 왔다. 특히 온실가스의 대부분을 차지하는 이산화탄소(CO2)의 대기 중 농도는 기하급수적인 증가세를 보이고 있다[2, 3]. 이러한 이산화탄소의 양을 줄이기 위해 대기로 배출되기 전에 고농도로 모은 후 압축 수송해 저장하는 기술인 탄소 포집 및 저장(carbon capture and storage, CCS) 기술은 가장 현실적인 방안으로 알려져 있으나 높은 에너지가 요구된다는 단점이 있다[4]. 따라서 CCS에 대한 대안으로 최근 이산화탄소를 원료로 이용하여 화학물질을 제조하는 연구가 활발히 진행되고 있다. 에폭사이드(epoxide)와 이산화탄소를 이용해 리튬이온전지의 전해액 등으로 널리 활용 가능한 산업적으로 중요한 물질인 고리형 카보네이트(cyclic carbonate)를 생성하는 반응에 대한 연구가 그 중에 하나이며 반응식은 Figure 1과 같다[5, 6]. 이 반응은 단점이 있는데 높은 활성화 에너지(activation energy)를 가지기 때문에(55~59 kcal/mol) 높은 발열(exothermic)에도 불구하고 비촉매 상태에서는 자발적으로 반응이 일어나지 않는다는 것이다[7]. 그로 인해 지난 수십 년간 포스핀[8-10], 유기염기[11], 이온성 액체[12-17], 유기금속착물[18-21], 금속산화물[22-24], 담지된 촉매[25-27] 등 다수의 균일 및 불균일 촉매들이 개발되어 왔다.
고리형 카보네이트 합성반응에 관한 연구는 주로 실험적인 방법론을 통해 다양한 촉매에 대하여 반응의 전환율(conversion)이나 선택도(selectivity)를 측정하게 된다. 하지만 최근에 본 연구팀에서는 앞서 언급하였던 계산화학을 이용하여 촉매가 관여된 고리형 카보네이트 합성반응의 이론적인 메커니즘을 제시하고 실험적으로 얻어진 결과를 해석하였다 - ZIF (zeolitic imidazolate framework) [28], 암모늄염(ammonium salt) [29], 금속 할로겐염(metal halide salt) [30], 알카놀아민(alkanolamine) [31]. 본 논문에서는 본 연구팀에 의해 타 문헌에 제시된 반응 메커니즘들을 새로이 에너지도(energetics)로 통합하여 정리하고 반응물과 생성물, 반응 중간체(intermediate)들의 에너지를 바탕으로 촉매반응 메커니즘에 대한 열역학적, 속도론적 해석을 수행하고자 한다. 또한 타 문헌에서 생략된 불균일 촉매를 다루는 자세한 계산방법을 소개함으로써 추후 유사한 연구를 수행하고자 하는 타 연구자들에게 도움을 주고자 한다.
2.1. 밀도범함수 이론(density functional theory, DFT)
계산화학(computational chemistry)이란 이론화학 또는 분자모델링(molecular modeling)이라고도 불리며 과거와 현재를 어우르는 학문 분야이다. 현재 양자화학과 컴퓨터의 지속적인 발전으로 인해 계산화학은 순수과학을 넘어서서 여러 분야에서 범용적으로 적용되고 있다. 분자모델링의 방법론 중에 본 연구에서 사용된 DFT는 부정확성과 많은 계산을 해결하기 위한 수단으로 전자 파동함수를 전자밀도로 대체시킨 이론을 말한다. 슈뢰딩거가 파동방정식에 대한 논문을 1925년에 출간했지만 전자가 1개인 H 원자의 경우를 제외하고는 그 방정식에 대해 해석적으로 정확히 풀 수 있는 해법은 없었다. 이 후 1998년에 Kohn은 DFT를 제시하였는데 이는 각각의 전자들의 파동함수로 전자들의 위치를 파악하는 것이 아니라 전자들을 전자밀도라는 하나의 집합으로 생각한 것이다. 즉 슈뢰딩거 방정식의 경우 전자의 수가 많아질수록 복잡함이 증가되지만 DFT는 상대적으로 훨씬 더 간단하게 다뤄질 수 있음을 알 수 있다. 이러한 Hohenberg와 Kohn의 정리에 기초하여 Parr와 Yang에 의해 DFT는 분자의 에너지와 속성을 계산하기 위한 도구로써 “계산 DFT”로 명명되었다[32].
DFT는 최근 대부분의 분자 모델링 연구에서 주로 사용되고 있는데 분자 내부의 전자 분포, 에너지 등을 양자역학적으로 접근한다[33]. DFT는 실험 및 분석에서 도달한 결론의 유효성 검사를 하거나 또 다른 가능성을 도출할 수 있기 때문에 가치 있는 실험도구로 널리 알려져 있다. 또한 DFT를 이용한 적절한 계산은 반응이 일어날 때 반응물, 생성물 및 반응 중간체의 알맞은 분자 구조, 진동운동 주파수, 에너지 등을 정확하게 구분해 낼 수 있다. 따라서 이러한 DFT를 이용한 분자 특성 계산은 이론과 실험을 밀접하게 연결해 주며 이는 종종 결과 해석에 중요한 단서로서 활용된다[34]. 앞서 언급했듯이 DFT는 전자밀도를 다루기 때문에 경쟁 방법론으로 잘 알려져 있는 파동함수를 다루는 Hartree-Fock 이론에 비해 꽤 정확한 결과를 빠르게 도출할 수 있다는 장점이 있다[35]. 물론 신뢰성을 향상시키기 위해서 DFT를 적용할 때 계산과 실제 측정값을 비교하여 오차범위를 판단하는 것이 필요하다.
모든 계산은 Gaussian 09 프로그램을 사용하여 수행하였다[36]. 구조 최적화를 시키는 범함수(model chemistry)로는 가장 일반적으로 사용되는 B3LYP법을 사용하였다. B3LYP란 잘 알려진 Becke potential에 Lee, Yang, Parr에 의해서 3개의 매개변수를 보완한 hybrid 모델을 의미한다[37-39]. 밀도함수를 구하기 위해 쓰이는 기저집합(basis set)은 원소에 따라 다른 값을 사용하였다. 일반적으로 큰 기저집합일수록 더 정확한 분자궤도함수를 얻을 수 있지만 계산 시간이 많이 소요된다. 본 연구에서는 6-31g(d)와 6-311++g(d)와 같은 가우시안(gaussian) 형태를 가진 적절한 크기의 기저집합을 사용하였다. 기저집합 6-31g(d)은 내부의 원자 껍질(shell)에 속한 전자궤도함수를 6개의 단일 가우시안으로 수학적으로 구현한다는 것을 의미하며, 최외각 전자궤도함수를 두 개의 기저함수로 표현하는데 3개의 primitive 가우시안으로 첫 번째 기저함수를 수학적으로 구현하며 두 번째 기저함수를 단일 가우시안으로 표현한다는 것을 나타낸다. 따라서 1이 하나인 31은 double zeta valence 기저집합을, 1이 2개인 311은 여기서 더 확장된 triple split valence 기저집합을 의미한다. 그리고 6-311++g(d)에서 +는 diffuse function을 의미하는 기호로 분자 결합으로 인해 궤도함수가 원래보다 넓게 퍼지는 경우 사용하며 이는 진동운동 주파수에 대해 6-31g(d)보다 높은 정확도를 가지고 있다[40]. 반면 많은 전자를 가지고 있는 금속과 같은 원소의 경우 최외각 전자들만을 고려하고 나머지는 하나의 core로 묶어 계산해 주는 effective core potential (ECP)을 이용하는데 본 연구에서는 잘 알려져 있는 LanL2DZ (Los Alamos National Laboratory double zeta) ECP를 사용하였다[41]. 즉, 요오드(I) 원소에 대해 LanL2DZ를 사용하였고 나머지 원소는 상황에 따라 위에서 언급한 6-31g(d)와 6-311++G(d,p)를 사용하였다. Gaussian 09 프로그램에 의해 구조 최적화가 이루어진 반응물, 생성물 및 반응 중간체 분자들의 경우 조화 진동수(harmonic frequency) 계산에 의해 모두 에너지적으로 지역최소값(local minima)에 위치함을 확인하였으며 전이상태 분자들의 경우는 반응 좌표(reaction coordinate)에 해당하는 음수 값의 진동수가 존재함을, 즉 안점(saddle point)이 존재함을 확인하였다.
본 연구에서는 Figure 1에 제시된 바와 같은 에폭사이드와 이산화탄소에 의해 오원환 카보네이트이 합성되는 반응이 촉매로 인해 어떠한 영향을 받는지를 계산화학으로 설명하고자 한다. 이 반응은 대부분 에폭사이드가 용매로 작용하기 때문에 별도의 용매 없이 진행되며 때로는 생성물이 용매로 작용하기도 한다. 따라서 유기 용제로 인한 폐수가 발생하지 않아 친환경적인 공정이라고 할 수 있으며, 별다른 정제과정 없이 비교적 간단한 공정을 거치기 때문에 경제적인 이점도 있다[42]. 앞서 언급했듯이 55~59 kcal/mol에 달하는 높은 활성화 에너지를 극복하기 위하여 촉매를 사용할 수 밖에 없는데 지금까지 본 연구팀에 의해서 보고되었던 네 가지 촉매 계에 계산화학 결과를 다음과 같이 정리하고자 한다. 참고로 계산화학에 앞서 우선 해야 하는 것은 반응경로를 예측하는 것인데 이는 일반적인 유기화학의 지식에 근거하여 이루어진다. 즉, 반응물, 생성물 및 반응 중간체의 가능한 초기 구조(initial geometry)를 예측하고 에너지적으로 해당 분자의 국소 최소값을 가지는 구조를 계산화학에 의해 최적화한다. 다음 전이상태 분자의 초기 구조는 그렇게 얻어진 반응물, 생성물 및 반응 중간체의 최적화 구조를 바탕으로 예측하고 마찬가지로 계산화학에 의해 안점에 해당하는 구조를 찾아간다.
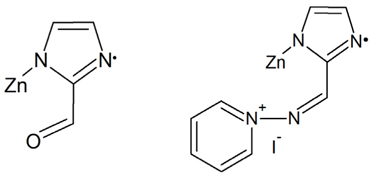
Zeolitic imidazolate frameworks (ZIFs)는 metal organic frameworks (MOFs)와 유사한 물질로 최근 생겨난 새로운 종류로 금속이 가운데 위치하고 있고 유기 이미다졸레이트들이 연결된 제올라이트의 구조를 띠는 입체구조를 가진 물질이다. ZIF라고 부르는 이유는 zeolite의 Si-O-Si에 해당하는 145°의 배위각도를 MOF의 metal-imidazolate-metal (M-IM-M)의 결합 각도에 적용시켰기 때문이다[28]. ZIF가 가진 이러한 구조는 기존 MOF의 높은 열 안정성과 영구세공도, 높은 표면적을 가지고 있을 뿐만 아니라 MOF보다도 훨씬 뛰어난 화학적 안정성을 가지게 한다. 특히 각종 화학공정의 부산물인 이산화탄소의 경우 ZIF와의 친화력이 높은 것으로 알려져 있다[43]. 이산화탄소와의 친화력은 ZIF의 화학적 및 열적 안정성이 높은 특성을 살려 이산화탄소 포집에 많은 응용을 할 수 있게 한다[44]. ZIF-90은 결정성 분자로서 3차원 구조를 가지며 zinc를 중심으로 하여 imidazole-2-carboxaldehyde (ICA) 링크로 이루어진 물질이다. Zinc nitrate tetrahydrate (Zn(NO3)24H2O)와 H-ICA를 이용하여 용매열합성법(solvothermal method), 기상확산법(vapor diffusion method) 등의 방법으로 합성되며, 합성된 ZIF-90의 분자식은 Zn(C4H3N2O)2이다[45].
ZIF-90의 경우 Bloch 이론과 반복경계조건(periodic boundary condition)을 사용하여 3차원의 전체구조를 고려한 고체계산을 진행할 수도 있지만 본 연구에서는 계산 시간을 크게 단축하기 위하여 알데하이드 작용기가 달린 ZIF-90의 최소 반복구조를 떼어내어 이를 기반으로 계산을 진행하는 cluster approximation을 사용하였다[46-49]. 따라서 한쪽 질소 원자가 비공유 전자를 가지므로 전체 가상 분자의 spin multiplicity는 doublet으로 설정하였고 기저집합도 계산 시간을 단축하기 위하여 6-31g(d)를 사용하였다. 비교대상인 IL-ZIF-90는 aminopyridine iodide를 ZIF-90에 고정화시켜 요오드(I)가 음전하를, imine 부분이 양전하를 띠는 이온성 액체(ionic liquid)가 ZIF-90에 담지된 구조로 볼 수 있다. 따라서 ZIF-90에서와 같이 최소 반복구조에 대해 spin multiplicity를 doublet으로 설정하고 기저집합도 동일한 6-31g(d)를 사용하였다. 두 경우 모두 가상 분자 전체의 전하(charge)는 중성(neutral)인데 다만 IL-ZIF-90의 경우 촉매반응에 참여할 때 요오드 음이온(I−)이 자유롭게 움직일 수 있는 장점이 있다. Figure 2는 계산에 사용된 ZIF-90과 IL-ZIF-90의 최소 반복구조를 보여준다. 그리고 계산에 사용한 에폭사이드는 같은 계열에서 분자의 크기가 가장 작은 PO (propylene oxide)였다.
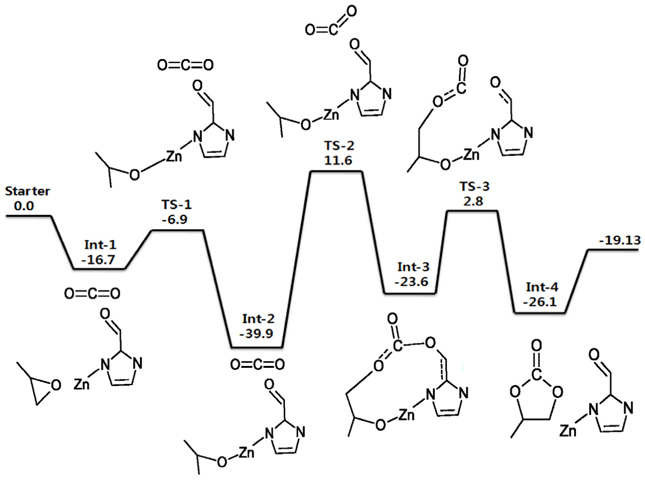
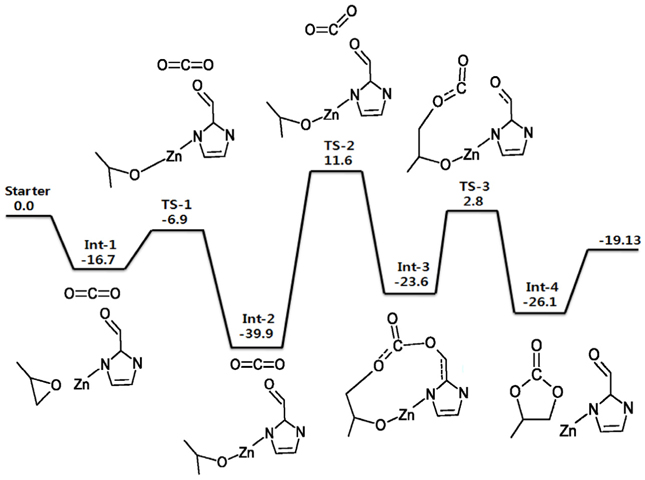
위에서 언급한 ZIF-90과 IL-ZIF-90 촉매에 대하여 에폭사이드와 이산화탄소가 반응하여 고리형 카보네이트가 생성되는 반응경로를 Figure 3과 4에 각각 에너지도로 나타내었다. 우선 두 반응경로의 가장 큰 활성화 에너지 값들이 51.5와 56.47 kcal/mol로 촉매가 없는 Figure 1의 반응(촉매가 없는 반응)의 실험적으로 알려진 활성화 에너지인 55~59 kcal/mol과 거의 비슷하였다. 촉매를 사용할 경우 활성화 에너지가 크게 낮아진다는 통념에 어긋나는 것으로 보이지만 이는 계산이 전체 3차원의 ZIF-90이나 IL-ZIF-90의 고체 구조에 대한 것이 아닌 반복되는 최소의 부분만을 고려한 cluster approximation에 의해 진행되었기 때문이다. 즉, 클러스터(cluster)가 일반적인 singlet의 안정한 분자 구조가 아닌 불안정한 doublet의 가상분자 구조이므로 절대적인 에너지 값에 큰 의미를 둘 수 없다. 다만 동일한 조건이므로 ZIF-90과 IL-ZIF-90의 반응경로의 에너지도는 서로 비교가 가능하며 실험적으로 구해진 전환율의 차이를 에너지도를 분석함으로써 이론적 근거를 제시할 수 있다.
전체적인 반응을 비교해보자면 ZIF-90 과 IL-ZIF-90의 반응은 Figure 3과 4에서 보는 것처럼 서로 다른 에너지도를 갖는데, 첫 번째 차이점은 속도 결정 단계(가장 높은 활성화 에너지를 가지는 단계)이다. ZIF-90의 경우 CO2의 화학적 삽입이 일어나는 Int-2에서 Int-3로의 반응이 속도 결정 단계인 반면, IL-ZIF-90의 경우는 Int-1에서 Int-2로의 PO의 고리 열림(ring opening) 반응이 속도 결정 단계이다. 두 속도 결정 단계에서의 에너지 장벽(energy barrier)은 ZIF-90와 IL-ZIF-90이 각각 51.2 kcal/mol, 56.47 kcal/mol로 서로 비슷하다고 볼 수 있다. 두 번째 차이점은 반응 중간체의 에너지 상태이다. ZIF-90의 경우 반응의 중간지점인 Int-2가 가장 안정적인 상태인 반면 IL-ZIF-90의 경우는 반응의 마지막 단계인 Int-5가 가장 안정적인 상태이다. 선례 연구에 따르면 PO 18.1 mmol을 120℃에서 3시간 반응시키면 IL-ZIF-90의 전환율은 97%, ZIF-90의 전환율은 51%로 IL-ZIF-90이 ZIF-90보다 더 높은 전환율을 나타낸다[28, 50]. Figure 4의 전체 에너지도에서 보듯이 IL-ZIF-90의 경우 에너지 장벽을 넘어서 정반응으로 진행되던 반응이 Int-5에서 PC (propylene carbonate)가 촉매로부터 분리되는 마지막 단계로 계속 진행되고 높은 에너지 장벽을 갖는 역반응으로 진행되지 않는다. 반면 Figure 3의 에너지도에서 보듯이 Int-2의 에너지 상태가 가장 안정되었으므로 Int-3로 진행된 반응이 역반응에 의해 다시 Int-2로 되돌아 올 가능성이 높다. 즉, 화학적으로 삽입되었던 CO2가 반응 중간에 다시 원상태로 분리될 수 있으므로 IL-ZIF-90에 비해 정반응으로의 계속된 진행에 의한 전환율이 상대적으로 낮아질 수 있다. 한편 Figure 4에서 요오드 음이온의 위치가 매우 자유롭게 형성되는 것을 알 수 있는데 이는 계산에 삽입된 요오드 음이온이 구조 최적화 과정에서 한 위치에서 다른 멀리 떨어진 위치로 움직인다고 생각하기 보다는 이온성 액체의 성질을 고려하면 해당 위치의 요오드 음이온이 사라지고 다른 떨어진 위치에서 다른 요오드 음이온이 삽입되는 것으로 간주하면 된다. 즉, 많은 요오드 음이온들이 주위에 존재하여 요오드 음이온이 각 단계마다 다양한 위치에서 반응에 참여하는 것으로 이해할 수 있다.
3.2. 폴리스티렌(polystyrene)으로 지지된 quaternized 암모늄염 촉매
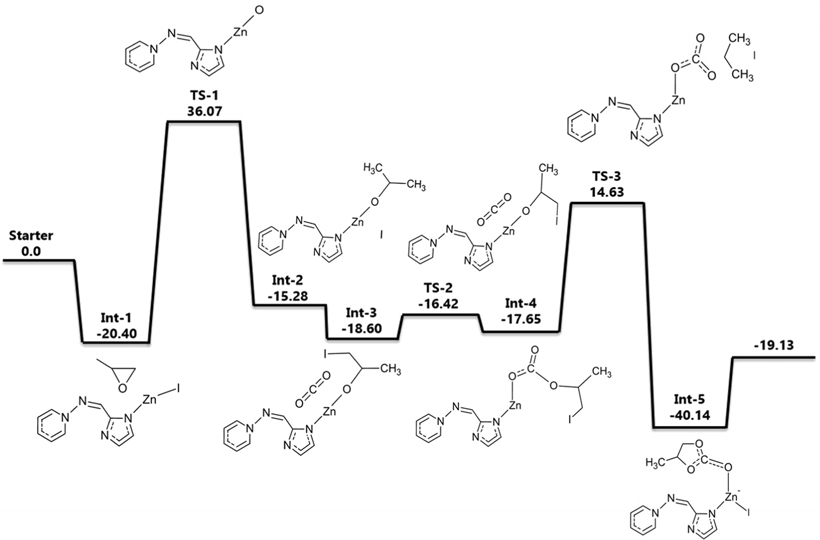
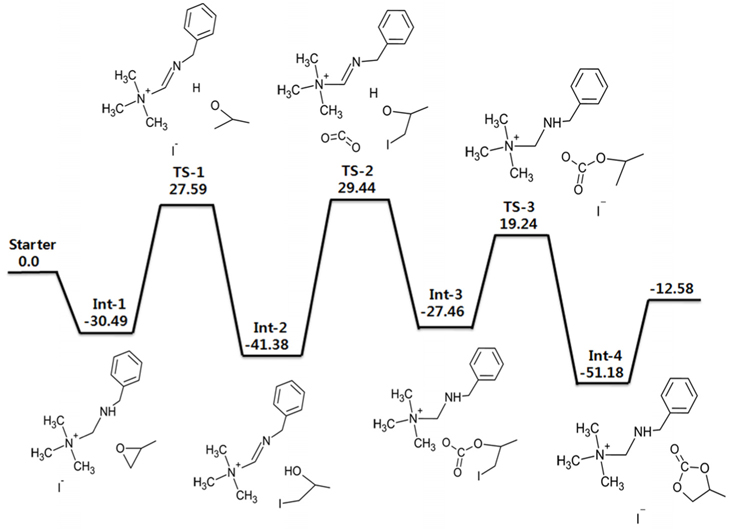
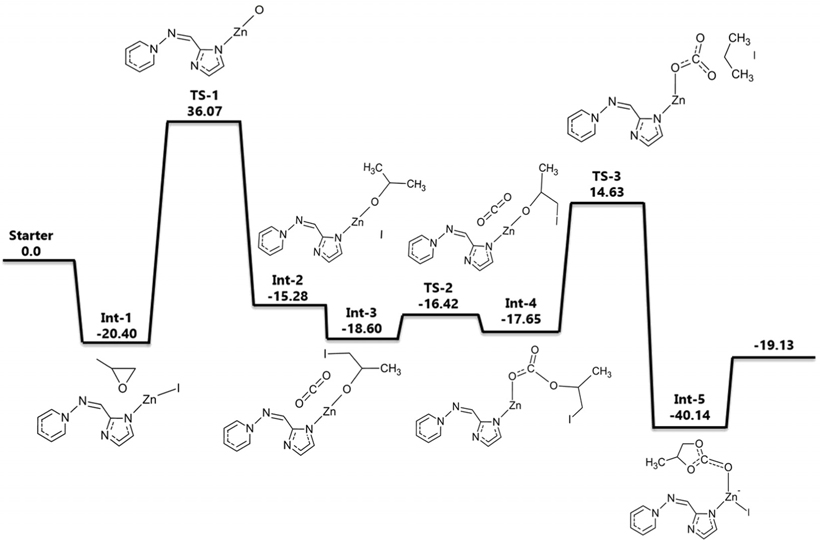
폴리스티렌에 담지시킨 quaternized 암모늄염을 촉매로 사용하였을 경우에 대해 고리형 카보네이트를 생성하는 반응을 계산하였다. ZIF-90에서와 마찬가지로 폴리스티렌은 무한히 반복되는 폴리머 체인(chain) 구조이므로 효율적인 계산을 위해 폴리스티렌 단량체만을 고려하여 말단기가 암모늄염으로 기능화(functionalized)된 가상 분자 구조를 기초로 삼았다. 비공유 전자가 없기 때문에 spin multiplicity는 singlet으로 설정하였다. 기저집합으로는 I 원소를 제외한 다른 원소들에 대해서는 6-311++G(d,p)를 사용하였고, I 원소에 대해서는 LanL2DZ를 사용하였다. 계산한 에너지 값은 에너지도로 Figure 5에 나타내었는데 가장 높은 에너지 장벽이 70 kcal/mol에 달할 정도로 Figure 1의 반응(촉매가 없는 반응)의 실험적으로 알려진 활성화 에너지인 55~59 kcal/mol보다도 높았다. ZIF-90에서와 마찬가지로 이는 폴리스티렌의 전체 폴리머 구조를 고려하지 못하고 반복되는 최소의 부분만을 고려한 cluster approximation에 의해 계산이 진행되었기 때문이다.
Figure 5에 제시된 에너지도를 보면 첫 번째로 PO가 촉매와 만나면서 형성된 중간체가 Int-1이며 에너지는 −30.49 kcal/mol이다. PO의 O 원자가 NH기의 H 원자와 상호작용을 하게 되면서 PO의 고리가 끊어지게 되는데 이것이 첫 번째 전이 상태(TS-1)이다. PO의 O-CH2결합이 끊어지게 되면서 친핵성(nucleophilic)인 I 원자가 C 원자를 공격하게 되는데 이때 C-I결합이 형성되면서 Int-2상태가 된다. 이 때 NH기의 H 원자는 PO의 O 원자와 결합을 형성한다. 그 다음 CO2가 들어오면서 PO와 상호작용을 하게 되는데 이때 두 번째 전이 상태(TS-2)가 나타난다. 이때 H 원자가 다시 NH기와 상호작용을 일으키게 된다. 들어온 CO2의 C 원자와 PO의 O 원자가 결합을 하고 O 원자에서 떨어져 나온 H 원자는 다시 NH기와 결합을 하면서 Int-3가 형성된다. 다음으로 C 원자와 I 원자의 거리가 멀어지면서 결합이 끊어지게 되고 O 원자와 결합하였던 CO2가 I 원자와 끊어진 C 원자와 상호작용을 일으키게 된다. 이것이 마지막 전이 상태(TS-3)이다. 다음단계에서 C-O결합이 이루어지면서 고리화 반응이 완결되는데 이 때가 마지막 중간체인 Int-4 (−51.18 kcal/mol)이다. 이러한 메커니즘을 통해 PC가 생성됨을 제시하였다. 이 반응에서도 이온성 액체와 같은 효과로 I 원자가 다양한 위치에서 자유롭게 반응에 참여할 수 있음을 알 수 있다.
본 계산의 선례연구에서는 에폭사이드 대신 AGE (allyl glycidyl ether)와 이산화탄소를 이용하여 고리화 반응을 진행하였는데, 120 ℃, 1.2 MPa의 조건에서 98%의 높은 수율을 보였다[29]. 선례연구와 달리 본 계산에서는 AGE대신 에폭사이드를 사용하여 간단한 계산을 실행하였는데, 계산결과 암모늄염의 −NH기와 에폭사이드의 O 원자 사이의 상호작용의 가능성을 확인하였고 선례연구에서 나타난 바와 같이 폴리스티렌에 담지된 암모늄염 촉매가 고리형 카보네이트 합성반응에 참여하는 메카니즘을 제시할 수 있었다. 앞의 ZIF-90의 경우와 마찬가지로 안정화된 폴리스티렌 폴리머의 전체 구조를 고려하지 못했기 때문에 정량적인 활성화 에너지에 의미를 두는 것보다는 가능한 반응경로를 제시하는 것이 목적이었다.
본 연구에서는 3.1과 3.2절과 같이 필요에 따라 클러스터 가정을 사용하였다. 이는 낮은 계산능력(computational capacity) 하에서도 비슷한 촉매 계에 대해서는 상대 비교를 통해 실험적으로 얻어진 상호 활성 차이를 해석할 수 있음을 보여주었다. 클러스터 가정은 주로 고체 촉매가 관여되는 불균일 반응(heterogeneous reaction)에 사용되는데 가정을 배제하고 Block 이론을 사용한 고체 계산을 수행한다면 불균일 촉매 계들 사이의 활성을 상호 비교하는 것이 가능하고 촉매를 스크리닝(screening)에 활용될 수 있다. 마찬가지로 다음 3.3과 3.4절과 같이 균일 반응(homogeneous reaction) 촉매 계의 경우 특별한 가정이 없이 계산이 가능하고 추가적으로 용매(solvent) 효과를 고려해 주면 균일 촉매 계들 사이의 상호 비교가 가능하다. 하지만 일반적으로 균일 촉매와 불균일 촉매에서 얻어진 계산화학 결과를 직접적으로 비교하는 것은 무리가 있다.
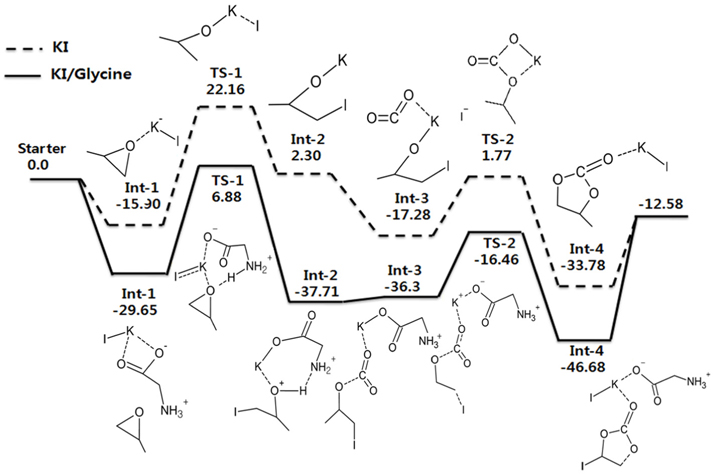
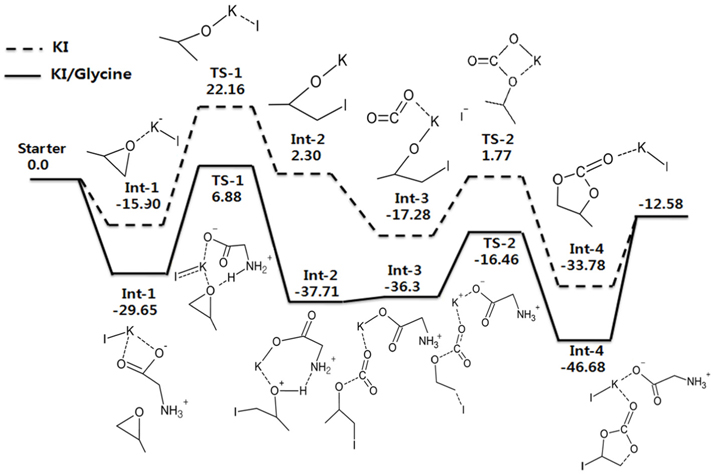
KI를 단독 촉매로 사용하였을 경우와 KI와 글리신(glycine)을 함께 촉매로 사용한 경우에 대하여 각각 계산한 반응 에너지 값인 에너지도를 비교하여 Figure 6에 나타내었다. 글리신은 대표적인 아미노산 조 촉매로 선정되었으며, 그 이유는 단순함과 글리신이 가진 쌍성이온의 특성 때문이다. 기저집합은 마찬가지로 I 원소를 제외한 다른 원소들에 경우는 6-311++G(d,p)를 사용하였고, I 원소에 대해서는 LanL2DZ를 사용하였다. Figure 6의 Path A는 KI 홀로 촉매로 쓰인 반응이며 Path B는 KI와 함께 조촉매로써 글리신을 사용한 반응을 나타낸다.
이 두 반응에서 먼저 주목할 것은 Path A와 Path B의 경우 모두 가장 높은 에너지 장벽이 각각 38.06 kcal/mol과 36.53 kcal/mol로 앞서 4.1절과 4.2절에서와 Figure 1의 반응(촉매가 없는 반응)의 실험적으로 알려진 활성화 에너지인 55~59 kcal/mol보다 훨씬 낮은 일반적으로 촉매가 활성화 에너지를 낮추어 주는 효과를 분명하게 확인할 수 있다. 그러나 두 에너지 장벽의 차이는 1.53 kcal/mol도 크게 차이가 나지 않아 에너지 장벽만으로는 두 반응의 차이를 얘기하기 힘들다. 따라서 우리는 Int-2의 에너지 위치에 관심을 가지고자 한다. Int-2 이후는 CO2가 화학적으로 삽입되는 반응이 이어지는데, Path A에서는 TS-1과 Int-2의 에너지 차이(역반응의 에너지 장벽)가 작기 때문에 Int-2가 CO2와 만나 후속 반응이 진행되기 전에 자발적으로 역반응이 일어날 수 있다. 반면 KI/글리신을 사용한 Path B에서는 KI 단독촉매를 사용하였을 때보다 Int-2의 에너지가 크게 낮아 역반응의 에너지 장벽이 44.59 kcal/mol임을 알 수 있다. 이는 Int-2가 현저히 안정됨으로써, 역반응의 가능성보다는 CO2와의 후속반응이 원활하게 진행될 수 있음을 의미한다. 즉, 염기성 아미노산의 조촉매가 반응에서 큰 상호작용을 발생시키는 것을 확인할 수 있었다.
본 계산의 선례연구에 따르면 120 ℃, 1 MPa의 조건에서 3시간 동안 반응을 진행한 경우 KI 촉매만 사용하였을 때는 3%의 낮은 수율을 보였지만 KI와 글리신을 함께 촉매로 사용한 경우 28%의 수율로 꽤 높진 않지만 KI만 사용하였을 때 보다는 높은 수율을 가짐을 확인할 수 있었다[30]. 이는 계산 결과와 같이 금속 할로겐염은 조촉매 없이는 촉매효율이 떨어진다는 사실과 일치한다. 따라서 본 연구가 선례연구의 실험을 뒷받침하는 이론적 근거가 됨을 알 수 있다.
3.4. Dimethylethanolamine (DMEA) 촉매
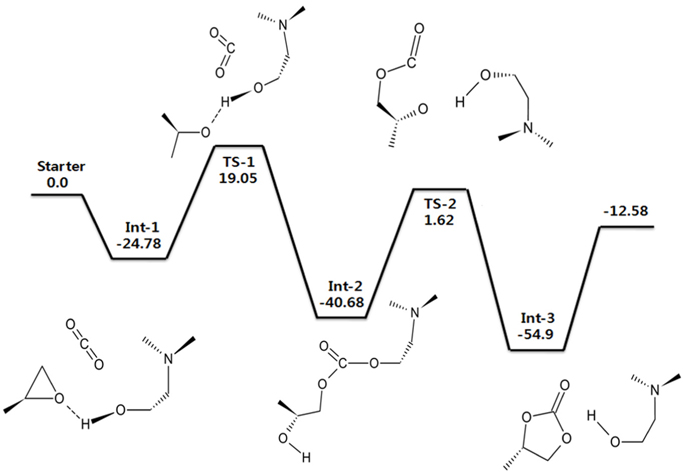
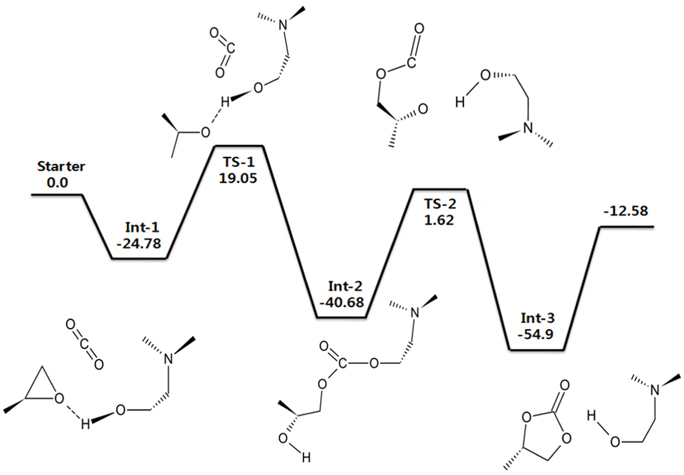
DMEA을 촉매로 사용하였을 경우에 대한 에너지도를 Figure 7에 나타내었다. 기저집합으로는 6-311++G(d,p)을 사용하였다. CO2와 PO가 촉매와 만나면서 Int-1을 형성하게 되고, 곧이어 알카놀아민에 붙어 있는 −OH기의 H 원자와 PO의 O 원자가 상호작용을 일으키면서 PO의 고리가 끊어지게 된다. 이때 첫 번째 전이 상태(TS-1)가 나타나게 된다. 곧이어 CO2가 들어오게 되는데 알카놀아민의 H가 떨어져 나간 자리에 CO2가 결합하게 된다. CO2의 분자가 굽어진 형태로 바뀌면서 알카놀아민과 PO와 결합을 하게 되는데 이때가 두 번째 중간 체인 Int-2이다. 이후 PO의 O 원자와 결합하였던 H 원자가 다시 알카놀아민의 O와 상호작용이 일어나면서 알카놀아민과 CO2의 결합이 끊어지게 된다. 상호작용이 일어나는 과정이 두 번째 전이 상태(TS-2)가 된다. 이때의 에너지는 1.62 kcal/mol로 높지 않은 에너지를 가지며 곧이어 나타나는 Int-3는 −54.9 kcal/mol로 매우 안정적인 반응임을 알 수 있다. 또한 Int-3에서는 오환원 카보네이트가 생성되는 것을 확인할 수 있다. 본 계산의 선례연구에 따르면 알카놀아민의 경우 금속과 할로겐염이 포함되지 않은 새로운 촉매로 선정된 촉매라고 한다. 또한 실험결과는 DMEA 촉매의 경우 82%의 높은 수율을 가지고 있는 것으로 나타났다[31]. 본 연구의 계산결과, 알카놀아민을 사용하였을 경우 에폭사이드와 이산화탄소의 반응과정에서 안정된 중간체를 가지며 활성화 에너지가 ~ 42 kcal/mol로서 촉매를 사용하지 않은 고리형 카보네이트 합성반응의 활성화 에너지인 55 ~ 59 kcal/mol와 비교하여 알카놀아민이 촉매로서 좋을 활성을 보여줌을 이론적으로 뒷받침할 수 있었다
본 연구는 분자모델링을 이용하여 에폭사이드와 이산화탄소를 이용해 고리형 카보네이트를 생성하는 반응에 대해 실험적으로 증명된 여러 가지 촉매의 활성을 이론적인 반응 메카니즘과 에너지도 탐색을 통하여 증명하는 것을 목표로 하였다.
1) 알데하이드로 기능화된 ZIF-90 촉매와 aminopyridine iodide를 고정화한 IL-ZIF-90 촉매는 계산의 편의나 시간 단축을 위해서 최소 반복구조(cluster)만을 떼어내 이를 유사 분자로 가정하여 계산하였다. 두 촉매의 반응을 비교해보면 속도결정단계와 가장 안정한 반응 중간체의 에너지가 다름을 알 수 있었으며 특히 후차의 차이에 의해 IL-ZIF-90이 ZIF-90보다 더 큰 전환율을 가지는 이유를 확인해 볼 수 있었다.
2) 폴리스티렌(polystyrene)으로 지지된 quaternized 암모늄염 촉매의 경우 ZIF-90과 마찬가지로 전체 폴리머 구조가 아닌 폴리스티렌의 단량체만을 고려하여 계산을 진행하였으며 암모늄염의 −NH기와 에폭사이드의 O 원자 사이의 상호작용의 가능성을 확인할 수 있는 반응경로를 제시하였다.
3) KI 촉매를 단독으로 사용하였을 경우와 조촉매인 글리신을 같이 사용하였을 경우를 비교해 본 결과 가장 높은 활성화 에너지는 크게 차이가 나지 않았지만 후자의 경우 중간 반응체가 훨씬 안정화되는 반응경로가 존재함을 확인하였고 따라서 반응이 정반응 쪽으로 유리하게 진행되어 실제 실험에서 얻어진 상대적으로 크게 향상된 전환율을 설명할 수 있었다.
4) 알카놀아민을 촉매로 사용하였을 경우 활성화 에너지를 ~42 kcal/mol로 낮춰줌으로써 금속이나 할로겐염 없이도 에폭사이드와 이산화탄소의 반응에서 촉매의 활성이 잘 일어남을 증명하였다.